第五课 运输者
存在于细胞膜中的运输、通道和泵是保持离子在细胞中平衡的关键,是为从神经到组织传播信号所不可缺少的。
通道和运输中造成缺陷的结果是各不相同的,取决於通道和运输的位置在哪里及输送什么。在心脏,缺乏钾离子通道就不能传导电子脉冲,导致在长QT综合症中看到心律不齐。在肺中,如不能在上皮细胞中输送钠和氯离子,就会导致囊细胞钎维化充血,这是最常见的一种遗传缺陷。彭德莱综合症看上去与硫酸盐输送缺陷有关。
第一节 囊扦维化
囊纤维化(CF)在今天的美国,是一种最常见的致命遗传性疾病,它使患者产生一种阻塞肺的厚而粘的肌肉,容易引起感染并出现终止在小肠起消化作用的胰腺酶阻滞。
CF是由缺陷基因引起的,它编码于钠和氯(盐)输送者,该输送者是在肺和其它器官界线的上皮细胞中发现的。在这种基因中找到几百个突变,所有这些突变通过上皮细胞产生钠、氯运输缺陷。CF临床表现的严重性直接与患者遗传特殊突变的特点有关。
由于在1989年发现CFTR,所以CF研究明显加快,1990年,科学家成功地克隆了正常的基因并在实验室把它加入到CF细胞中,校正钠、氯运输缺陷机制。这种基因治疗的技术在有限的患者中作了试验。然而,这种治疗并没有象原先期望的那么有效,在采用基因治疗之前,还需作进一步研究。同时,其它的实治疗也应证明有用于与CF的斗争。

第二节
听觉损失是相当普遍并可出现在从婴儿到老年的任何年令段。大约1%的婴儿患有重度听力障碍,其中一半是先天遗传,存在许多耳聋基因。而且在美国和欧洲人口中,引起听力障碍的最普遍原因是连结蛋白26(Cx26)基因的突变。与囊纤维化一样,Cx26有3%携带率,它引起大约20%的儿童耳聋。
Cx26突变引起先天性综合症,非综合症的耳聋不会伴有如视觉缺损这类的症状。Cx26被定位于染色体13q11-12并编码称为连结蛋白26的间隙结合蛋白,间隙结合蛋白是原生质膜通道,它让小分子和离子在毗邻细胞之间运动。内耳的间隙结合蛋`白可能起保持钾离子体内平衡的作用,它对内耳的功能和听力是重要的。已认识到Cx26的突变可能破坏钾离子循环,因而出现耳聋。
发现Cx26突变是引起先天性听力损失,能帮助早期诊断听力损伤。耳聋早期诊断和治疗对语言和社会交际的开发是重要的。

第三节 畸形发育
畸形发育异常(DTD)是一种罕见的生长障碍,患有该种障碍的病人通常有短而畸形足、变形手关节。该疾病虽然在所有人口都能发现,但在Finland中尤为普遍。
发生在DTD中的基因突变定位于染色体5并编码一种新奇的硫酸盐运输者。它与DTD患者在不同组织中观察到的不寻常硫酸盐浓度相匹配。因为作为关节减震器的软骨,在它制造过程中需要硫酸盐,所以硫酸盐对骨关节是重要的。在软骨中添加负电荷有助于减震作用。
在充分认识医疗条件和开发有效的医疗方法前,必须进一步做大量的研究。

第四节 血友病A
血友病A是一种遗传性血液疾病,主要影响男性,血友病是以缺乏凝血蛋白因子VIII,使患者产生不正常渗血。Babylonian Jewe 在1700年前首先叙述了血友病,当王后Victoria把该病介绍给欧洲几个皇室家庭后才引起公众广泛注意。在X染色体上HEMA基因的突变引起血友病A。正常情况下,女性有两个X染色体,而男性有一个X染色体一个Y染色体,因为男性仅在X染色体有任何基因的一个拷贝,他们不能象女性那样增加一个基因拷贝而消除危险。
因此,象血友病A这类与X-染色体有关的疾病在男性中更为普遍,凝血因子VIII的HEMA编码基因主要在肝脏中合成,它是与血凝结相关的许多因素之一,即便其它凝结因子仍然存在,只要凝血因子VIII单独缺损就足以引起血友病。

第五节 LONG-QT综合症
LONG-QT综合症(LQTS)起因于心脏钾离子通道的不正常结构,使受到影响的人感染加速心率失常。这就会导致突然丧失意识和引起突然的心脏病患者死亡。患有心脏病的青少年当他面对从锻炼至大声喧嚷的压力都会出现心脏瘁死的危险。
LQTS通常是一种常规染色体显性遗传疾病,定位于染色体11上的LQT1发生突变,导致自己心脏钾离子通道发生严重缺陷,不能把电子传导脉冲至整个心脏。还可能存在其它基因,不确定地定位于染色体3、6和11,它的突变产物可能导致或引发LQT综合症。
Beta封闭剂常用于治疗疾病综合症,并对某些综合症病人产生效果。然而,普遍有效的治疗方法,比如避免激烈的体育锻炼和其它压力仍是有效的。有关上述讨论基因如何相互作用的研究应为LONG-QT治疗加速新的开发。

第六节 门克氏综合症
门克氏综合症是一种标志细胞吸收铜能力减弱的先天性代谢缺陷,特点为大脑严重变性及动脉病变,导致在婴儿期死亡。头发稀疏发脆并在显微镜下呈卷缩状。
门克氏疾病是患者不能输送铜的X连锁隐性性状遗传疾病,输送铜需要与制造骨胳、神经和其它结构相关的酶来执行。包括I型埃勒斯-当洛斯综合症在内的许多其它疾病,可能是等位基因突变的结果(例如,在同一基因上发生突变,但稍有不同的临床症状),有望对这些疾病的深入研究可有效用于对门克氏疾病的斗争。
如果在刚出生的头几个月里就加强治疗,铜组氨酸盐对某些患者呈现出增加生命的有效预期性。然而,这种治疗增加生命只能预期从三岁到十三岁,所以只能认为是一种缓冲作用。在鼠模型中存在相同的门克氏病的条件,对这类鼠模型的研究将能帮助阐明人类铜输送的机制,为门克氏患者开发有效的治疗方法。

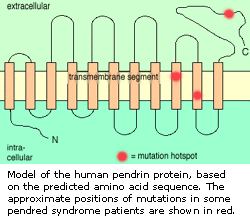
第七节 彭德莱综合症
彭德莱综和症是造成10%遗传性耳聋的一种遗传性疾病,表现为先天性双侧神经性聋,伴甲状腺肿形成,而无甲状腺功能减退。最近发现的彭德莱综合症遗传病因为科学家一百多年的困惑作出了解释。
1997年十二月,在NIN’s国家人类基因组研究机构,使用物理图谱来帮助缺认可能引起彭德莱综合症的异常基因。
正常基因标识一种Pendrin蛋白质,它只有在甲状腺有效水平中找到,且与大量硫酸盐输送密切相关。但这种蛋白质的基因发生突变时携带这种突变基因的患者将在临床上显示出彭德莱综合症。
因为甲状腺肿不总是只在患彭德莱综合症的病人身上找到,缺陷的Pendrin基因弄清楚原来与某些耳聋有关,而在次以前没有认识到这点。Pendrin的发现还激发起把新的研究手段用于甲状腺研究,并改变硫酸盐输送在人类疾病中的作用。
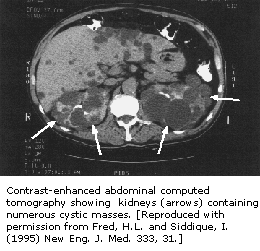
第八节 成人多囊肾脏病
成人多囊肾脏疾病(APKD)是以在一个或两个肾脏中出现大囊、导致慢性肾衰竭为特怔的疾病。肾脏在人体中作用是,过滤血液中以尿和细胞外流动的浓缩氢、钠、钾、磷及其它离子形式出现的代谢最终产物。
1994年欧洲多囊肾脏病协会从染色体16中分离出一种基因,染色体16在具有APCD的家属中遭到破坏。被PKD1基因编码的蛋白质是一种包含细胞-细胞间反应、细胞-基质间反应的整体薄膜蛋白。PKD1在正常细胞中的作用可能与中间微管功能有关,如在薄中放置Na(+),K(+)—ATPase
离子泵。程序性的细胞死亡或细胞凋亡可能也包含在APKD中,进一步搞清楚疾病的发病机制期待做进一步研究。
所以,称为“CPK”鼠是人类疾病众所周知的模型。期待对病鼠分子基础的究为提供更好理解人类疾病并希望出现更有效的医疗效果。
第九节 镰状细胞性贫血
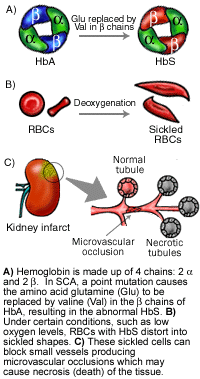
镰状细胞性贫血(SCA)在美国是最常见的遗传性血液疾病,大约有72000个美国人受到影响或在非洲后裔美国人中的概率为1/500。SCA是以发生疼痛、慢性溶血性贫血、和严重感染为特征的疾病。通常开始于儿童期。
SCA是一种常规染色体隐性疾病,在染色体11p15.4发现的血红蛋白Beta基因突变引起贫血。携带HBB的频率在世界各地不同,由于携带者有点儿抗疟疾能力,所以高发率与高发疟疾地区有关。
大约8%非洲后裔美籍人是携带者。HBB的突变导致产生结构不正常的Hbs血红蛋白(Hb),Hb是一种产生血蛋白的携氧蛋白,具带色特点。在某些如低氧或高浓度血红蛋白的条件下,具有Hbs血红蛋白的患者,其不正常的Hbs簇聚集,把受扰乱的RBCs变成镰刀形,改形并固定下来,结果就产生疼痛甚至形成危害性组织。
虽然到目前为止,SCA还没有治愈的方法,但流质、止痛药、抗体、和输血的结合常用于临床治疗。一种抗肿瘤药羟基脲在防止疼痛危象方面已显示出效果。当患SCA的患者服用羟基脲时,诱导胎儿形成Hb(HbF),使胎儿期或新生期产生的正常aHb,防止镰刀型贫血产生。已开发出的SCA鼠模型,正在成为有效治疗SCA的潜在新方法。
第十节 肝豆状核变性
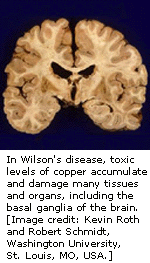
肝豆状核变性是一种罕见的铜输送常规染色体隐性遗传疾病,由于产生铜的类积使毒素进入肝和脑,肝病是儿童中最常见的临床症状,神经性疾病在青年中是最常见的。眼睛角膜也受到影响:凯澤尔—弗莱舍尔环使角膜周围带深铜色环,它表示铜的沉积。
肝豆状核变性疾病(ATP7B)被定位于染色体13上,发现基因的顺序与由铜输送所引起的另一种门克氏疾病基因缺陷部分相似。编码束缚铜区域的相同顺序,它是与门克氏病蛋白非常相似的P-型ATPase越膜泵的一部分。
人类ATP7B基因的同源性已被定位于鼠的第八条染色体上,人类疾病在鼠中的可靠模型也是有用的(称为Long-Evens Cinamon[LEC]rat),这些系数有利于研究铜的输送和肝病理生理学,将帮助开发治疗肝豆状核变性疾病的方法。
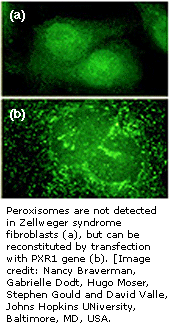
第十一节 澤尔韦格综合症
澤尔韦格综合症是一种影响儿童并能引起死亡的罕见遗传性疾病,异常问题在产前就发生了,肿大的肝、血液中高水平的铁和铜及视觉障碍是澤尔韦格综合症的主要临床表现。
被定位于染色体12的PXR1基因,发生突变就引起澤尔韦格综合症。PXR1基因是在动物细胞,尤其在它们肝、肾和脑细胞过氧化物酶体-微体的表面发现的受体产物,虽然过氧化物酶体参与许多代谢方面的重要反应,但它的功能还不完全清楚。PXR1受体为把酶输入过氧化酶体所必需,只有它功能发挥,过氧化酶体才能使酶发挥它们的重要功能,比如,细胞脂质代谢和代谢氧化作用。
酵母菌与人类有同原的PXR1,它提供有力的分子遗传技术,将被用于细胞中过氧化酶体正常功能及发生在疾病状态分字水平的研究。