第四课 肌肉和骨胳
骨骼提供附着肌腱、能产生力量的肌肉抗基点。许多疾病是由对肌肉形成、功能起重要作用并连结组织的基因发生缺陷所引起的。(连结组织是一个包括骨骼、软骨、和腱在内的大课题)。
原纤维缺陷――是在产生强而韧性的组织中起重要作用的连结组织蛋白质---引起马方综合症(DMD),而骨骼畸形是在软骨中缺乏硫酸盐输送者所引起的。
肌肉细胞本身缺陷所造成的二种原发性疾病是杜兴(型)肌营养不良(DMD)和肌强直营养不良(DM),DM是另一种“动力突变”疾病。亨廷登舞蹈病也同样如此,它与肌肉蛋白质激酶基因发生突变造成核甘酸酶重复扩展有关。DMD与缺乏对维持细胞结构起重要作用的细胞支架蛋白、肌细胞增强蛋白有关。
而埃―克综合症基因已被定位。我们期待把蛋白质功能理解为疾病的基础。
第一节 杜兴型肌营养不良
杜兴型肌营养不良(DMD)是以肌肉假肥大性为特证的一组肌肉营养不良的疾病。DMD是一种最流行的肌肉营养不良症,并以肌肉快速退化为特征的疾病,它发生在生命的早期阶段,全部是伴性的,主要影响男性,大约在世界范围内的发病率为1/3500男孩。
在X-染色体上发现了DMD的基因,编码一个大的肌细胞蛋白。在肌肉细胞中为维持细胞结构支架需要肌细胞蛋白,可想象为通过把细胞支架内部成分固定至细胞膜表面来加强肌细胞。一旦失去了肌细胞蛋白,细胞膜就变得可渗透,细胞外面的组分就会进入细胞内,增加细胞内压直至肌细胞“爆炸“和死亡,随后的免疫应答就发生危险。
已有一种DMD的鼠模型,它有利于进一步论证我们对肌细胞蛋白正常功能和疾病途径的理解。尤其是,为弥补病鼠肌细胞蛋白的损失增加肌细胞蛋白生产的初步实验有希望成功。

第二节 埃-克综合症
也可叫做“软骨外胚层发育异常”的埃-克综合症是以短肢、侏儒症、多指(多脚趾,或多或少手指)、腕骨畸形、指甲营养障碍、部分唇裂、心脏变形和经常产前长牙为特证的罕见遗传性疾病。
引起埃-克综合症EVA的基因已被定位于染色4的短臂上。到目前为止,对健康EVA基因的功能还不清楚,自从提出了该疾病的分子机理,这就成为要回答这种疾病的最重要的问题之一。
埃-克综合症经常在Lancaster County Pennsylvania的老年辈Amish community中找到。因为这组人群小而孤立的,所以能得到难得的机会去观察这一特殊人群一代又一代的情况。所观察到的遗传途径已指出这是隐性常规染色体疾病。(例如,获得双亲基因突变后,才对疾病的影响变得明显。)

第三节 马方综合症
马方综合症是一种结缔组织疾病,所以影响许多结构,包括骨胳、肺、心脏、眼睛和血管。该疾病通常以细长肢为特征,据信已影响Abrahama Lincoln.
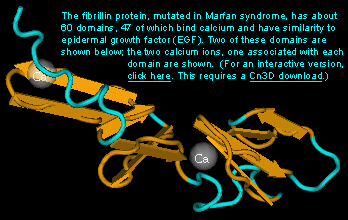
马方综合症是一种被定位到染色体15上FBN1基因的显性常规染色体疾病。FBN1编码一种称之为原纤维蛋白,它是形成结缔组织弹性纤维的基础。没有这种原纤维提供的结构支持,许多组织是纤弱的,其严重性也由此而来,例如主动脉壁破裂。
Beta封闭剂常用于控制某些马方综合症的心血管症状,然而,它们不能有效对付骨胳和眼睛疾患,这些疾患还很严重。在模型鼠中鼠原纤维合成、分泌物和结缔组织形成的研究将对进一步掌握人类马方综合症是有希望的。
第四节 肌强直症
肌强直症是一种肌肉收缩而力量减弱至松弛。具有这些症状后,肌肉也变软弱无力并消耗了。肌强直症能引起智力缺陷、头发脱落和白内障。这种罕见的疾病一般发生在年青人中,然而它能发生在不同的年令阶段并具有极为不同的危险程度。
在第19条染色体上找到肌强直基因,给骨肌肉中发现的蛋白激酶编码,这种基因编码可能起调节作用。
这种疾病的普遍特怔是它的症状随着每一代的出身变得更严重,这是因为一代一代基因错误复制导致基因组“AGC/CTG三连体重复”扩大,同样的情况发生在亨廷病上。在5至27个的AGC/CTG复制拷贝之间不会对个体产生影响,至少需50个重复拷贝才会对肌强直病人产生影响,而影响更为严重的病人扩大至几千碱基对。

第五节 脊柱肌肉的萎缩
以脊柱运动神经元的死亡和随后的肌肉瘫痪为特征的脊柱肌肉萎缩(SMA)是一种引起儿童致命的最普通遗传神经肌肉疾病。SMA发生的年令和严重性从由呼吸衰竭引起早期死亡的婴发生期(I型)到减少预期寿命(2型)、不能行走(2型、3型)的青少年温和发生期各不相同。
阐明疾病的原因认为,在染色体5上找到生存运动神经元基因(SMN1)的等位基因发生突变,使SMA成为隐性常规体疾病。典型解释SMN1缺损引起突变,或者也是位于染色体5上,称为SMN2的几乎同一基因取代了SMN1。SMN1和SMN2都给同一蛋白编码,但SMN1基因产生全尺寸的蛋白质,而SMN2基因产生截短的编码蛋白和少量的全尺寸蛋白,SMN1基因都缺损引起I型SMN,而当SMN1被SMN2取代增加SMN2的复制数量时,产生青少年型病。影响个体的SMN2基因数越多,全长的蛋白将产生越多,就产生温和的疾病形式。已知在产生mRNA中起关键作用的编码蛋白质SMN1和SMN2通过男孩表达出来,但只能在脊柱运抵神经元的特别高水平时才能找到。
SMN1的功能目前通过鼠和转基因鼠在进行研究,这种蛋白和它的功能的特征甚至将导致在与帮助控制SMN发病危险性的基因治疗结合应用中充满希望。
