��ʮ���¡�ҽԺҩѧ�ල
����ҩƷ�ල������ָ�������������������չ��ҷ��ɶ�ҩƷ��������Ӫ��ҵ��ҽԺ���е�λ�й�������������˽��еĹ�����ҽԺҩѧ�ල������ҩƷ�ල��������ɲ��֡�����Ҫ�����ǣ�����ִ᳹�С�ҩƷ����������ҩ�����棬�ڽ���������������ҩƷ�ල������ͬʱ��������������ҽԺҩ�������취���ȷ��棬��ҽԺ�ڲ�ʵʩ��Ч�ļල����֤��ҩ��ȫ��Ч�����ҽ���������ٽ�ҽ����ҵ��չ��
����һ��ҩѧ�ල������������
������ҩƷ���������᳹ʵʩʹҩƷ�ල������Ϊ��һ�����˵Ĺ���ѧ�ơ�ҩƷ�ල�����漰��ҽѧ��ҩѧ����ѧ��ʷѧ������ѧ�Լ������������ִ���ҵ�����������ں������Ӷ�����õ����ط�չ��Ŀǰ���ҹ��Ѿ��γ����ԡ�ҩƷ��������Ϊ�����ҩ��������ϵ��ͨ������GMP��GSP��GLP�������淶����ʹҩƷ��������Ӫ��ҵ���ƻ����淶��������������ҩ������һϵ����������������ʹ������ҩ���У�Ϊ�����β��ṩ�˸�����ֶΡ��������ҹ�����ҩѧѧ���о�ˮƽ��ߡ���ҽԺҩѧ���棬���������ɵ�һ���ٴ�ҩƷ������Ӧ��չ����ҩ�ٴ��о�������ҩƷ��ѡ��ҩƷ��̭��ҩ�ﲻ����Ӧ��⣬������ҩ��ҽ����ҩ�����Լ�ҩѧ��������Ϣ������������ݡ�ҽԺҩѧ�ķ��ٷ�չ����ҽԺ�������ã�����ְ�𣬹���������ҽҩ��Ա��֪ʶ�ṹ��������µ�Ҫ��
����ҽԺҩѧ�ල������������ҩƷ������ҩƷ��������Ʒ�������Ƽ��������У�ҩƷͻ���ر����˲���������������ԣ�ҩƷ��������ҽ��ָ����ʹ�á���֮�õ����ﵽ���μ�����Ŀ�ģ���֮����ʧ֮��������Σ����ᣬ������������ҩԴ�Լ�����ҩ��IJ�����Ӧ������Ե���ɵ����Σ����ҩƷ��Ӧ���뼰ʱ�ɿ��������ٴ���Ҫ��ҩƷӦ��ȫ��Ч��ȷ�����������������������ۣ�ʹ�ü���ҩƷ��ҩƷ������Ϲ���ҩƷ�����������뾭����ϸ����ʹ�ã�ҩƷ���������ʾ�����ҽԺҩѧ�ල�������ۺϹ������ԡ������˶�ҩƷ���ڵ��������й������������Ӱ��ҩƷ������������ء����˽��мල���������˵Ĺ����������мල������
����������������г������ƶȵ�Ѹ�ٽ����ͷ�չ�����ù�����ΪҽԺ��������Ҫ֧�����ӹ������л�����ҩĿ¼������ҽ��ҩƷ����Ŀ¼��ҽ�Ʊ��գ��Լ���ֹҩƷ������лؿۣ���ҽԺ�����ҹ�ҩ������ҩ����������ҩ�����ٴ�ҽ����ҩ������ҽԺҩ���Ƶľ��ú������������Ԥ�㡢�ɹ�����������⡢���⡢���䡢��Ӧ�����㡢ͳ�Ƶ����ʹ����������������ù����ķ��룬���ֶ���ҽԺҩѧ�ල���������ݡ��ӹ����ĽǶȹ���ҽԺҩѧ�ල�����ʣ�����ҩƷ��������Ϊ�������Ա�֤ҩƷ����Ϊ���ģ��ԡ��ˡ������Ϣ������ʱЧ���������й������������ִ������Ļ���Ҫ�ء�����ҩƷ�ල������Ŀ���DZ���������ҩ��ȫ��Ч��ά���������彡��������������Ч�档ҽԺҩѧ�ල������Ŀ���DZ�֤ҩƷ�����������ٴ���Ҫ������ʹ��ҩƷ�����ҽ��ˮƽ��ҽԺҩѧ�ල�����������ִ����������ۡ������������ԡ�ҩƷ���������Լ�������ҽԺҩ�������취�ȷ��桢������Ϊ�����ı��������ල��ϵ�����Ƹ��ֹ����ƶȣ���ҽѧҩѧ�Ļ�������Ҫ�أ��ˡ��ơ����Ϣ������ʵʩ������ʵʩ��ѧ���淶������������������С�ľ���Ͷ�룬������ľ���Ч�棬ͬʱ��֤ҩƷ����������ҩƷ��Ч������������ҩ��ȫ��Ч��
��������ҩѧ�ල����֯������ְ��
������һ��ҽԺҩѧ�ල������ְ����������ҽԺҩ�������취����һ������������涨��ҽԺҩ���Ƹ���Ժҩ����������Ժ��ֱ���쵼�£����ա�ҩƷ������������ʵʩϸ��ල����鱾Ժ��ҽ�ƿ��Һ���ʹ��ҩƷ����ֹ���ú��˷ѡ���ҽԺҩ�������취���ڶ��µ������涨��ΪЭ����ָ��ȫԺ������ҩ�Ϳ�ѧ������������ҽԺ�����أ�Ҫ����ҩ��ίԱ�����Ҫ�����ǣ���Ժ��ҩ�ƻ����ƣ��ޣ�����Ժ������ҩĿ¼�ʹ����ֲ��˱�Ժ���Ƽ�����֯��������ҩ����ٴ���Ч�벻����Ӧ�������̭Ʒ���������ʱ�о������Ժҽ����ҩ�ش�����ල��鱾Ժ�ִ᳹��ҩ�������ִ���������ҽԺҩ�������취����ҩ���ƿƵ���֯�������й涨ҩ����Ҫ��ǿҩƷ����������������ȫҩƷ�ල�ͼ���ƶȣ��Ա�֤�ٴ���ҩ��ȫ��Ч��ҽԺҩ�¹���ίԱ����ҽԺҩѧ�ල������������Ŧ��ҽԺҩ������ҽԺҩѧ�ල����֯ʵʩ�ߡ�
���������������������ŵ�ҩƷ�ල����������ְ��
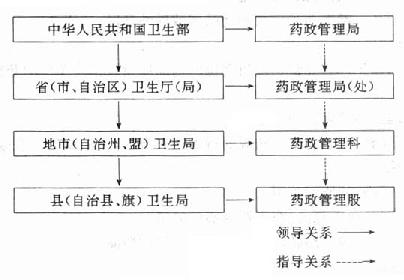
����1.�ල����������ͼ15-1��
ͼ15-1�ҹ�ҩƷ�ල��������ʾ��ͼ
������ҩƷ����������һ�µڶ����涨��������Ժ����������������ȫ��ҩƷ�ල������������
�������ҹ�����������ҩ�������֣���ʡ����������ֱϽ�е���������������ҩ�������֣������������������ݣ�����������������ҩ���ơ�����Ͻ���ڵ�ҩƷ�ල����������
����ҩ�¹����Ķ���������ҩƷ�������ĺ�����ҩƷ������������Ŀ����ȷ��ҩƷ��ȫ��Ч��
���������������ŵ�ҩ����ҩ�������ͬ��ҩƷ��������Ӫ��ʹ�õ�λ��ҩƷ���������ͼ����������Ϊ����ֻ�Ա���λ��ҩƷ������������ҵ�ڲ��Ĺ������������������ŵ�ҩ���������Ǵ������Ҷ�ҩƷ���мල��������ִ�вþ��ĵ�λ��
����2���ල����������ְ��
������1��������ҩ�������ֵ�ְ��Χ
������ִ�С�ҩƷ�����������ⶨʵʩϸ�ල�����ִ᳹����������������ҩ���䲼������ҩ֤�顱�����ĺš�
����������ҩ����������ҩ����������
�����ܼල����ҩƷ����̭�������ô���Ч��ȷ�е�ҩƷ��
�����ݹ�������ҩƷ������ҩƷ������ҩƷ�ͷ�����ҩƷ����ҩƷ��֤��
���������������ҩƷ�ල�������������Ž�����ҩƷ��֤��
�����ල���ҩƷ��������Ӫ��ʹ�õ�λ��ҩƷ�����Լ�ҽ�Ƶ�λ��ҩ��������
�����ฺ��ҩƷ����������
��������顢����ҩƷ�������ж����¹ʡ�
�������Υ����ҩƷ���������ü��йط������Ϊ�����Σ����д���������Ҫ��Ū�����εģ���˾�����ſظ档
������2��ʡ��ֱϽ�С����������������֣�����ҩ�������֣���ְ��Χ��
���������ⶨ��ҩƷ��������ʵʩϸ���������ҩ���䷢��ҩ֤���⣬����������ְ����ͬ�����⣬������˷����Ƽ�����֤������ҩƷ��Ӫ��ҵ����֤������ҩƷ������ҵ����֤����
������3��ȡ����ҩƷ
�����ټල����顶ҩƷ���������ڱ��ص�ʵʩ�����
�����ڹ�������ҩƷ������ҩƷ������ҩƷ�ͷ�����ҩƷ��
������ȡ����ҩƷ
�����ܼල���ҩƷ��������Ӫ��ʹ�õ�λ��ҩƷ������ҽ�Ƶ�λ��ҩ��������
�����ݼලҩƷ������
�������顢����ҩƷ�������ж��¹ʡ�
�����߶�Υ����ҩƷ�����������йط������Ϊ�����Σ����д�����������������ʱ����˾����������ظ档
������4���أ��죩ҩ���ɵ�ְ��Χ
�����ټල����顶ҩƷ���������ڱ�Ͻ��ʵʩ�������
�����ڹ�������ҩƷ������ҩƷ������ҩƷ�ͷ�����ҩƷ��
������ȡ����ҩƷ��
�����ܼල���ҩƷ��������Ӫ��ʹ�õ�λ��ҩƷ��������ҽԺ�Ƽ�������
�����ݼලҩƷ������
�������顢����ҩƷ�������ж��¹ʡ�
�����߶�Υ����ҩƷ�����������йط������Ϊ�����Σ����д�����
����������ҩƷ����������ְ��Ͻ�����֣��ҹ���ҩ˨����Ϊ�����������й�ҩƷ������Ʒ�춨����ʡ����������ֱϽ��ҩƷ���������أ��С��ݡ��ˣ�ҩƷ���������أ��죩ҩƷ��������
������ֹ1986�꣬�ҹ�ҩƷ��������Ѵ�1400������ڱ���Ա��15000���ˣ��������γ������������ͼ15-2��
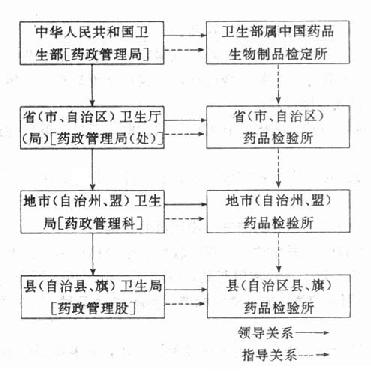
ͼ15-2�ҹ�ҩƷ�������ʾ��ͼ
����1�����������й�ҩƷ������Ʒ�춨���й�ҩƷ������Ʒ�춨����ȫ��ҩ��������ҵָ�����ĸ���ҩ�������ϼ�ҩ����ָ����
�����й�ҩƷ������Ʒ�춨������ȫ����ҩƷ�����ල������ͼ����ٲù������мƻ�����֯ȫ����ҩƷ���顣�μӡ��й�ҩ�䡷��������ҩƷ�������ⶩ������������ҩ�������ˣ�ҩƷ�����ù��ұ�ҩƷ���о����궨�ַ��ȹ�������֯�ⶨȫ��ҩ���ѧ�������ˣ�ҩƷ�����ù��ұ�ҩƷ���о����궨�ַ��ȹ�������֯�ⶨȫ��ҩ���ѧ������չ�滮���ٰ�������͵�ҩ��רҵ���ް���ҩ���鱨�����ȡ�
����2��ʡ��ֱϽ�С�������ҩ�������𱾵���ҩƷ�����ල�����顢�ٲù������Ե���ҩ����ҽҩ��Ӫ��ҽ�Ƶ�λ��ҩƷ����������飬����ҩƷ������̬���ⶩ�����ط�ҩƷ�����е��ϼ�ҩ���������ҽ����ҩƷ����ݡ���Ʒ�ı궨����ҩ�������˼���ҩƷ��������
����3���أ��С��ݡ��ˣ�ҩ�������𱾵���ҩƷ�����ල�����顢�ٲù������Ա�����ҽҩ�����������ˡ���飻����ҩƷ������飬����ҩƷ����������ලҽҩ��������Ӧ��ʹ�ò��ŵ�ҩƷ������ָ���أ��죩ҩ������ҵ������
����4���أ��죩ҩ���������ҩ������ͬ������ҩ����Ӧ��ҩ�¹�����ҩƷ�������������ص���ҩƷ�ල��
�������ģ�ҩƷ���л�����ҩ���ǹ��Ҷ�ҩƷ���������ͼ��鷽�������ļ����涨����ҩƷ���������顢������ʹ�õȵ�λ��ͬ���صķ������ݡ�����ʽ������ʹ�õ�ҩƷ�ͺ�ҩ���ƶ�ҩƷ�����Ա��˺���ҩƷ��������ҩƷ��Ӧ����ȷ��ҩƷ��ȫ��Ч�����鼼���Ƚ������ú��������鷽��Ҫ����ȷ����������㡢���ٵ�ԭ��Ҫ����ʵ����������Ҫ��ӳ�¼�����Ӧ�úͷ�չ��ע�����������Ƚ�������Ҫ����������������Ӱ�����������ء�����������·�߸ı䣬ԭ��������Ч�ؿ��Ƹò�Ʒ����ʱ��Ҫ����������ҩƷ���ƶ�������Ӧ�����˵����
��������ҩƷ�������ط�Χ��
����1���й�ҩ������ط�Χ���ط�������ģ���Ч�϶���������С����һ���ı��涨�ܿ��ƻ�춨������Ʒ�֡�
������1�����������������ճ��졢�����ȶ��ɹ�ҵ������ҩƷ��
������2��ҽ�Ƴ��ã�Ʒ����Դ������м�����α�ͱ�Ҫ�������涨����ҩ�ļ���Чȷ�С���Դ�ḻ�����г��죬���Ƽ��г��õIJ�ҩ��
������3��ʹ����㡢�������������ճ��졢ԭ����������г�ҩ��
������4�����ٴ�������鷽���Ƽ�������ѡ�ա�ҽ�Ƴ��÷��ϡ����ҵ�Ҳ�ʵ����ء�
����2������������ط�Χ
������1���й������ش�Ʒ�֡�����û���������ķ�����ҩƷ��������ҩƷ����ҩ�˹��ϳ�Ʒ������ҩƷ��
������2���ϰ�ҩ�����ع��������а汾δ����ġ���Ч�϶������ڼ�ʡ����������ʹ�ò���������ҩƷ��
������3����Ч�϶����������������һ���Ľ�����ҩ��
������4���ط������صģ�ҽ�Ƴ��á���Ч�Ϻá����������࣬��Ҫͳһ����Ʒ�֡�
����1963�겿�������102�ֳ���ҩƷ��1972�겿�俹���ر�����17�֡�1979�겿�������ҩƷ��ҽ��ͬλ�ء������صȱ�������43��Ʒ�֣�ͬʱ�䷢����ҩ�ı�������50��Ʒ�֡�
����1983����������һ����ǿҩƷ�������ƶ�������ȫ�깲�䲼����119����1984��ȫ��䲼�˼������������50��ҩƷ�IJ�����]
����������Ʒ����ǹ��Ҷ�������Ʒ�����������ͼ춨�Ļ���Ҫ��1977�꿪ʼ����������Ŀǰ�����ĸ���������Ʒ�����켰�춨��̽�����ϵͳ������������������1979��9���������������䷢�Ĺ�̹�110����
����3.�ط�ҩƷ�����صķ�Χ
������1���й�ҩ����δ���أ��ɹ�ҵ�����ģ���Ч�϶�����һ����Ч��������ʡ������������������ҩƷ��
������2��ҩ��Ͳ����δ���أ��������Ƚϳ��û�������ҩ�ģ�썵��������ص���Ʒ����Դδ�������������ϰ��Ӧ�ã���Ч�ڵĿɲ���Ϊ�ط�����
������3����ҩ����Ƭ������Ʒ�����ƶ����������ء�
������4���ٴ������鷽��ҽԺ�����Ƽ�����Чȷ�еĿ����ƶ���̣�����ҵ����������ط�����
����1982�꣬ȫ��27��ʡ�������ط�ҩƷ��978��������ԭ��ҩ90�֣���ҩ�Ƽ�383�֣���ҩ�Ƽ�505�֡��ط������ƶ���ҽҩ��չʷ����������������ã����ǣ����ڸ������յ���������һ��Ҳ������һ�£�ʹ��һЩ��ҩ�в�ͬ�ı���ͬһ��ҩ��ڼ�ʡ���ܱ���������ʡ�ɿ��ܱ�����������ȥ��ȫ���ĵط�ҩƷ���Ʊس��ֻ��Ҿ��档ͳһȫ���ط����Ĺ�����ʮ�ִ���������ڱ��С�1986�����������Ҫ��ȡ����ҩ�ط������ط�ϰ��ҩ��Ҫ���ƶ��ط���������ȡ���������ͳһ��������ġ�
�����������ɼල
���������������ҹ�������������彨��IJ�ͬʱ���Ⱥ�䲼���й�ҩƷ�����ķ�����������Դٽ��ҹ�ҽҩ��չ���˱�֤���á��ְ����ڹִ᳹�е���ҽ�Ƶ�λ��ҩ�����������йص���Ҫ����ժ�г�������������ҩ�Ƽ�������Ա���ģ�����ִ�С��Ծ������й�ҩƷ���ɼල�����ڱ�֤ҽԺҩѧ����������ҩƷ�������ż�Ϊ��Ҫ���塣
������һ�����л�����ҩƷ������������ơ�ҩƷ������������ȫ���˴�ί���������ߴλ���ͨ�����л�������ϯ��1984���18�������1985��7��1����ʩ�С������ҹ���ʷ�Ϲ��Ұ䲼�ĵ�һ��ҩ�����棬��ҩ������������һ����Ҫ���������ƶ���Ŀ����Ҫ���ҵ������й�ҩƷ�ල�������ߺ�ԭ���ù��ҷ��ɵ���ʽȷ����������ҩƷ���������ڹ��Һ������Ⱥ�ڵ��ϸ�ල֮�£���֤ҩƷ��������ҩƷ��Ч������������ҩ��ȫ��ά���������彡����ȫ�Ĺ�11��60�������±���Ϊ����һ�����ڶ���ҩƷ������ҵ�Ĺ�����������ҩƷ��Ӫ��ҵ�Ĺ�����������ҽ�Ƶ�λ��ҩ��������������ҩƷ�Ĺ�����������ҩƷ�İ�װ�ͷ�װ�����������������ҩƷ���ڰ���ҩƷ���̱���Ĺ������ھ���ҩƷ�ල����ʮ�·������Σ���ʮһ�¸���
�������������л�����ҩƷ������ʵʩ�취�������ʵʩ�취��������Ժ��������������1989���1�����ʩ�С����Ǹ��ݡ�ҩƷ�����������ܽ��ҹ�3������ҩƷ�������мල����ʵ������Ļ������ƶ�����Ҫ���档ȫ�Ĺ�10��57�������±���Ϊ����һ�����ڶ���ҩƷ�ල����ְ�𣻵��������������֤�ij���������ҩ��������������ҩƷ�����ĺţ�������ҩƷ������ҵ�Ĺ�����������ҩƷ��Ӫ��ҵ�Ĺ������ڰ���ҽ�Ƶ�λ��ҩ���������ھ��´�������ʮ�¸���
����������������ҩƷ�����취���ɹ���Ժ1987��11�·���ִ�С��Ǹ��ݡ�ҩƷ���������Ĺ涨��Ϊ�ϸ��������ҩƷ����֤ҽ�ơ���ѧ�����еİ�ȫʹ�ö��ƶ��ġ�ȫ�Ĺ�8��38�������±���Ϊ����һ�����ڶ�������ҩƷ���ָ˺�����������������ҩƷ�Ĺ�Ӧ������������ҩƷ�����䣻����������ҩƷ�Ľ����ڣ�����������ҩƷ��ʹ�ã������·��ڰ��¸���
�������ģ���ҽ���ö���ҩƷ�����취���ɹ���Ժ1988���23�����ʩ�С��Ǹ��ݡ�ҩƷ���������Ĺ涨��Ϊ�˼�ǿҽ���ö���ҩƷ�Ĺ�������ֹ�ж����������¹ʵķ������ƶ��Ĺ����취��ȫ�Ĺ�14����
�������壩������ҩƷ�����취���ɹ���Ժ1988���23�����ʩ���Ǹ��ݡ�ҩƷ���������Ĺ涨��Ϊ�˼�ǿ����ҩƷ�Ĺ������ƶ��ġ�ȫ�Ĺ�8��28�������±���Ϊ����һ�����ڶ��¾���ҩƷ�������������¾���ҩƷ�Ĺ�Ӧ�������¾���ҩƷ�����䣻�����¾���ҩƷ��ʹ�ã������¾���ҩƷ�Ľ����ڣ��ڰ��¸���
������������������ҩƷ�����취������Ժ1989���25�����ʩ�С��Ǹ��ݡ�ҩƷ���������Ĺ涨��Ϊ�˼�ǿ������ҩƷ�Ĺ������ƶ��ġ�ȫ�Ĺ�7��31�������±���Ϊ����һ�����ڶ��·�������ҩ�����ơ��ٴ��о��������������·�����ҩƷ����������Ӫ�ͽ����ڣ������·�����ҩƷ�İ�װ�����䣻�����·�����ҩƷ��ʹ�ã������·�����ҩƷ���ͼ��飻�����¸���
�������ߣ���ҽԺҩ�������취������Ժ1989��3�°䲼ִ�С����Ǹ��ݡ�ҩƷ���������Ĺ涨��Ϊ��ǿ��ҽԺҩ�������Ĺ������ƶ��ġ�ȫ�Ĺ�9��32�������±���Ϊ����һ�����ڶ���ҽԺҩ�¹���ίԱ�������ҩ���Ƶ���֯���������µ������Ƽ���������ҩ���ļ��飻������ҩƷ�Ĵ����빩Ӧ��������ҽԺҩ����ѧ�о����ڰ���ҩѧ��Ա����������ߣ��ھ��¸���
�������ˣ���ȫ��ҽԺ����������ҽԺ�����ƶ���ҽԺ������Աְ����������1982��䷢����������ҽԺ������Աְ������ȷ�涨��ҽԺ����������Աְ��Χ�����а�����ҩ������Ա��ְ��Χ���Լ�ǿ����������Ա�������ģ�ʵ�и�λ�����ƣ���ͬ����ҽԺ������
�����ġ�ҩƷ�����ල
�����ල�ǹ�����ְ��֮һ��ʵ���ϸ��ҩƷ�����ල���������ڱ�֤ҩƷ����������ҩƷ��Ч������������ҩ��ȫ��ά���������彡���Ⱦ��м�Ϊ��Ҫ�����塣
������һ��ҩƷ�����ĸ���ʲô�����������ʱ�����֯��ISO����������һ���������¶��壺��������ָ��Ʒ����ҵ���ߵĵġ������Լ������Ƿ�Ϻ��涨Ҫ���һ�����Ի����ܡ�����Ʒ����ָ���Dz�Ʒ�ܹ�������������ǵ������һ���������߱�����Щ��Ȼ�ڸĻ�����Ҳ����ʹ�ü�ֵ����Щ���������˲�ͬ��Ʒ�IJ�ͬ��;�����������ǵIJ�ͬ��Ҫ����Ʒ��ͬ����;���죬���ǶԲ�Ʒ������Ҫ��Ҳ��ͬ�����ǰ�����Ҫ���Ϊ��Ʒ���������ԡ�������һ����̬�ĸ����Ʒ���������Ե���Ҫ�̶Ȳ��ǹ̶�����ģ��⽫���û���Ʒ�����IJ�ͬҪ��������������ͬ���������ǣ���ע���á��ڷ��á���ҵ�ã����ǵ�����Ҫ���Dz�ͬ�ģ����������Ҳ�Dz�ͬ�ġ�
����ҩƷ������Ԥ�������ơ�����˵ļ�������Ŀ�ĵص����˵��������ܣ����涨����Ӧ֤���÷������������ʡ�����ҩƷ����;�Լ����������ҩ���飬ҩƷ���������Կ��Ը���Ϊ���¼��㣺��1����Чȷ�У���2��ʹ�ð�ȫ����������С����3���ȶ��Ժã���Ч�ڳ�����4����ҩ���㣻��5���۸���ˣ���6����װ�ʺϣ������ڴ��桢�����ҽ��ʹ�á�������Щ���������У�1���ͣ�2��Ϊ�ؼ��������ԣ���������������ԣ���Ϊ��û�����������ԣ��㲻�ܳ�ΪҩƷ����ˣ�����ҩƷ�������Ը���Ϊ��ȫ�ԡ���Ч�ԡ��ȶ��ԡ�
�������ſ�ѧ�����ķ�չ��ҩƷ���������������������ܿ�ѧ�ؽ������ȣ�Ҳ����ͨ��һϵ�����ݵ�ָ��ֱ�Ӽ�ӵط�Ӧ����������״���������顢������Ч�ۣ��ⶨ��pHֵ����ȫ���顢��Ѫ���顢���ʼ�顢����������ȣ���Ҫ��ͨ��������������ӳ�ģ��ѷ�ӦҩƷ�������Եļ���������ָ����ȷ�涨�������γɼ����ļ�����Ӧ��ҩƷ����������ҩƷ���������ɹ��һ��������ƶ����䲼����Ϊ����ҩƷ�������ҩƷ�������Ǽ��ҩƷ�Ƿ�ϸ�ij߶ȡ����Ҷ�ҩƷ�����ļල�������ԣ�ҩƷֻ�кϸ��벻�ϸ�֮�֣�ֻ�кϸ��ҩƷ����ʹ�á����������Ʒ��ͬ����Ϊ���������Ʒ�����е���Ʒ����Ʒ������Ʒ����ҩƷ�IJ��ϸ��Ǿ���������Ϊ��Ʒ������Ʒ�����������۵�ʹ�õģ��������ܵ������Ʋá�
����ҩƷ��������Ȼ�������ҩƷ����������������Ҫ��������ʹ�õķ�Ӧ���ر���������Ч��ȷ�С�������Ӧ������⣬�����о�ҩƷ�������������ԡ�
������ҩƷ������������ϵ������ҩƷ���������ۡ�ʹ�õȹ��������ĸ���ҩƷ���������ۡ�ʹ�õȹ���������ͬ��ҩƷ����������ֱ�ӷ�ӳҩƷ���������������Ƿ�ӳ���������ۡ�ʹ���б�֤ҩƷ�������Ĺ�������ˮƽ�����ۺϵط�ӳҩƷ��������Ӫ��ҵ��ҽ�Ƶ�λ�Ĺ���������������������֯�����ԴﵽҩƷ�������ı�֤�̶ȡ��Ӽ�ǿ����������Ҫ����˵�����������ҩƷ�������������ܣ�Ҳ�������ҩƷ���������ۡ�ʹ�õȹ���������
����������ҩƷ�����ල�����ĸ���ҩƷ�����ļල�������ǹ��������������Ÿ��ݹ���ͨ�������������Ȩ�������ݹ����ƶ���ҩƷ�������桢�ƶȡ����ߣ��Ա���ҩƷ���������ۡ�ʹ�õ�λ��ҩƷ�����Լ�Ӱ��ҩƷ�����Ĺ����������й����������Խ�����ҩƷ�����ļල������
����������ҩƷ�����ල�����ı�Ҫ�Ժ�����ҩƷ�����ල����ҩƷ������������Ҫ�Ծ����ģ���ҩƷ��ữ�������ı�Ҫ����ǿ�ѧ�����Ϳ�ѧ������չ�Ľ�������ı�Ҫ�Ժ����ã�������������Ϊ�����ϵ�ġ��������������¼��㣺
����1��ҩƷ���з��μ����������������ƻ���������Ҫ���á��������ҩƷ�־��в�ͬ�̶ȵĶ�����Ӧ����ר���Ժ�ǿ����֮�õ����β����ˣ���֮����������Σ��������ҩƷ����α���Ӻ��Ѵ����ֱ�����գ�ҩƷ����������ѧ�����Ժ�ǿ���ϸ�Ʒ�벻�ϸ�Ʒ������ҩƷ���������������������й�ϵ����Ʒ�����в��������Լٳ�����ı�Ա���������ҩƷ����Щ�����ԣ������ɹ����ƶ�ҩƷ�����ɴ������ҵ�ר�Ż���ʹ���ִ���ѧ���������÷����к������ķ�������ҩƷ�������мල���������п��ܱ�֤������ҩ��ȫ��Ч��
����2��ҩƷ�����;�Ӫ��ҵ�������������Ļ������ƶȣ�����������Ӫ������������������Ч������Ч�����ì��ʱ������ƫ���������;���Ч�档ֻ��ʵ�й��Ҷ�ҩƷ�����ල����������Э���Ϳ�������ì�ܣ���֤ҩƷ������
����3��ҩƷ�����ල�����Ǵٽ���ҩ��ҵ��ҽҩ��ҵ��չ�Ķ�������Ʒ�����ߵ��Ǻ������ҵľ��ü���ˮƽ����Ҫ��־����Ʒ���������ҩƷ��������Ӫ��ҵ��˵���ܷ���ںͷ�չ�Ĺؼ����⡣ҩƷ�����ල�����ƶȱؽ��ٽ���ҵ����ȫ����������������ҵ��ʶ�ͽ�����������е����⣬��ʹ��ҵ�ļ������졢�������£��Լ���߾�Ӫ����ˮƽ���ƶ���ҵ����ʡ�ط�չ�������ҵ�ľ���������
����4��ҩƷ�����ල�����ɼ�ʱ����ҩƷʹ���д��ڵ������������ٺ���̭��Щ������Ӧ����Ч��ȷ�е�Ʒ�֣�����ٴ���ҩˮƽ��
����5��ҩƷ�����ල��ά����ᷨ�͡��������ǽ���������������ͬ��Щ���������ۡ�ʹ�ü�ҩ����ҩ��Σ���������ı�ƺ�����Υ����Ϊ�����������
�������ģ�ҩƷ�����ල������ʽ������
����1��ҩƷ�����ල�������Դ����¼�������з��ࣺ
������1����ʱ��˳���ϣ�����Ϊ��ǰ�ල���ճ��ල���º�ල��
������2���Ӷ���Χ���棬���Է�Ϊȫ���һ��ල��ijһ�����ר�żල��
������3���Ӳ��÷�ʽ���棬���Է�Ϊ���Ҽල���ڲ��ල���ⲿ�ල��
������4��ҩƷƷ�ֵ����ٺ���̭��
������5����ҩƷ��������Ӫ��ҵ��ҽ�Ƶ�λ����ҩ���г���ҩƷ���м�顢���飬��ʱ����ҩƷ�������⡣
������6��ָ��ҩƷ������ҵ��ҩƷ��Ӫ��ҵ��ҩƷ�����������Ա��ҵ������
������7�����顢����ҩƷ�������ж��¹ʣ�ȡ��ҩ���������ϸ�ҩƷ��ִ����������������Ҫ���������ε���˾�����ſظ�ȡ�
�������壩ҩƷ�ලԱ�ƶ�ҩƷ�ලԱ�Ǹ��ݡ�ҩƷ���������Ĺ涨��������������������������ҩƷ�����ල��������
������ҩƷ���������й涨���ؼ���������������������ҩƷ�ලԱ��ҩƷ�ලԱ��ҩѧ������Ա���Σ���ͬ������������˷���֤�顣��ҩƷ�ලԱ��Ȩ���չ涨��Ͻʽ�ڵ�ҩƷ������ҵ��ҩƷ��Ӫ��ҵ��ҽ�Ƶ�λ��ҩƷ�������мල����顢���飬��Ҫʱ�����չ涨������Ʒ����ȡ�й����ϣ��йص�λ���þܾ���������ҩƷ�ලԱ��ҩƷ������ҵ�ͺͿ��е�λ�ṩ�ļ������ϣ������ܡ�
�����ҹ�����Щ���������ɹš������ȵ���ʵ�мලԱ�ƶȡ��ֽ�ҩƷ�ලԱ������һЩԭ���ԵĹ涨�������¡�
����1��Ŀ��ҩƷ�ලԱ�ǹ��Ҷ�ҩƷ������ʹ�ල����רҵ������Ա���ڸ������������쵼�¿�չ��������Ŀ�����ڼ�ǿ��ҩƷ�ļල��������ʹ���Ҹ����ְ����ά��������ҩ��ȫ��Ч��
����2��ҩƷ�ලԱ��������Ƹ���ҹ�ҩƷ�ලԱ��4����������ҩƷ�ලԱ��������Ƹ�β��䷢֤�飬��ȫ����Χ����ʹְȨ��ʡ����������ֱϽ�����������֣�ҩƷ�ලԱ��ʡ����������ֱϽ����������Ƹ�β��䷢֤�飬������������������ʡ����������ֱϽ��Ͻ������ʹְȨ���ء��С��ݡ��˼��أ��죩������ҩƷ�ලԱ�ɸ�����������Ƹ�β��䷢֤�飬��ʡ����������ֱϽ�������������ű������ڱ�Ͻ������ʹְȨ��
����ҩƷ�ලԱ��ҩѧ��Ա���Σ��ּ�ְ��רְ���֣�����4�꣬����Ƹ���Ρ�
����ҩƷ�ලԱ��ִ������ʱ�����б������ݵı�־��֤����
����3��ҩƷ�ලԱ��ְ��Χ
������1������Ƹ��Ȩ�ޣ���Ͻ����ҩƷ��������Ӫ��ʹ�õ�λ���г�ִ�С�ҩƷ����������ҩ��������мල��顣
������2������ȡ�á�ҩƷ������ҵ����֤����ҩƷ������ҵ���������������ҵ�����ա�ҩƷ���������������淶�����мල��飻�Գィ��Ľ���ҩƷ������ҵ���������䣩���ϡ�ҩƷ���������������淶����������м�����գ��Ա���֤��
������3����ҩƷ��Ӫ��ҵ���������������ҵ����ҩƷ���мල���ͶԳィ��ҩƷ��Ӫ��ҵ����֤��
������4����ҩƷ��Ӫ��ҵ���������������ҵ����ҩƷ���мල���ͶԳィ��ҩƷ��Ӫ��ҵ����֤��
������5����ó���г���ҩ�Ľ��мල��顣
������6���Խ���ҩ�ġ�ҩƷ���мල��顣
������7���Բμ����������ҵ�Ĺ��ң�����������й�ҩƷ������ҵ����ҩƷ�������졣
������8�����״ν��ڵ�ҩƷ����Ϊ��Ҫʱ���ɵ�ҩƷ����������������������ҵ�����������졣
������9�����������������ŵļƻ����Ż���ʱָ����й�����
������10���ල��顶ҩƷ�������������Ĺִ᳹�С�
����ҩƷ�ලԱ�ڽ��м�鹤��ʱ���������ü��ǰ���������������й����ϣ��ⶩ�����١�����Ӧ��ʱ���������������ύ��鱨�棬ͬʱ����ҩƷ��������
����4��ҩƷ�ලԱ��Ȩ��
��1)ҩƷ�ලԱ�а��չ���ҩƷ���������Ͻ���ڵ�ҩƷ��������Ӫ��ҵ��ʹ�õ�λ���г���ҩƷ�����мල����顢�����Ȩ����������2)ҩƷ�ලԱ��ִ������ʱ����Ȩ���鵥λ����ѯ�����������֤�鼰ԭʼ��¼���й����ϣ���Ҫʱ�ɰ��չ涨ƾ֤���������йص�λ����Ӧ������ϣ���ʵ��ӳ��������þܾ���������
����(3)ҩƷ�ලԱ�Լ�ҩ����ҩ�����ش����������ҩƷ����������ͣ��������ͣ���ۡ���ͣʹ�þ�����Ȩ������Ҫ��ʱ�������������ű��棬����һ���ĵ��鴦����
����(4)ҩƷ�ලԱ��Ȩ����˽�����ҩƷ������ҩƷ���������ҩƷ�����������ۡ�ʹ�����������ʱ��ӳ���ֵ��й����⡣
����(5)ҩƷ�ලԱ��Խ����ӳ�й�ҩƷ�����Ȩ����
����5��ҩƷ�ලԱ������
(1)�ڽ��й���ʱ��Ӧʵ�����ǣ�����ִ����������˽����������(2)����ѧϰҵ����Ϥ�йط��棬ģ�����ط��
����(3)�Ա���鵥λ�ṩ�ļ������ϣ�Ӧ�����Ʊ��ܣ����Ա��ܡ�
����6.ҩƷ�ලԱ��������ҩƷ�ලԱӦ�����Ρ�������ҩʦ������൱רҵ��������ˮƽ��ҩ����ҩ��ɲ�����ѡƸ�Σ�ʡ����������ֱϽ�����������֣�ҩƷ�ලԱӦ������ҩʦ���ϻ�����൱רҵ��������ˮƽ��ҩ����ҩ��ɲ�����ѡƸ�Σ��ء��С��ݡ��˼��أ��죩������ҩƷ�ලԱӦ��ҩʦ�����ҩ����ҩ�칤���������Ͼ���һ��ҩѧרҵ֪ʶ��ʵ�������ҩ����ҩ��ɲ�����ѡƸ�Ρ�
����ҩƷ�ලԱ����߱�����������
������1������ҩƷ��������Ӫ�����ܡ�ʹ�úͼ���ȷ��������֪ʶ��ʵ�����顣
������2����Ϥ������ҩ����ҩ���йط��롢���ߡ�����͡�ҩƷ���������������淶��֪ʶ��
������3������һ��ʱ���ҩƷ�ලҵ����ѵ�����������������ſ��˼���
(4)�������ɣ����ؼ��ɣ��Ȱ���ְ���������ػ��ܡ�����(5)���彡������ʤ�ι�����
����������Ⱥ���Ե�ҩƷ�ල�ලҩƷ����������Ӫ��ʹ�õ�λһ��˵�Ƚ����У����Դ����ģ����г���ƭǮ���˵���ҽҩ��������ҩƷ�ලԱ���У����뷢��Ⱥ�ڼල��ҩƷ��������Ӫ��ʹ�õ�λ�ĺܶ�Υ����ҩƷ������������ĻҲ��Ҫ�ڲ��ġ�֪���ˡ����ܱ��ҷ���ҩƷ�ලԱ�ͽҷ�����Ϊ��ӳ�˲�����Ϊ������ܱ���������Թ�ͱ�������ˣ������ƶ����ߣ�����ҩƷ�ලԱ�ͽҷ��ˣ��������ǵ�ÿһ�γɼ����������ͬ����Ҳ�����ֹҩƷ�ලԱ�ܻߡ��ൺ�н�������ҽԺҩѧ��Ա���ٴ�ҽ����Ա�У�Ƹ�μ�ְҩƷ�ලԱ����ҩƷ�����ල���˺ܺõ����á�
�����塢ҩƷ�İ�ȫ����Ч�Լල
����ҩƷ�ල�����ĺ�����ҩƷ��������Ϊ�˱���������ҩ��ȫ��Ч����ô����ȫ����Ч֮��Ĺ�ϵ����أ�
��һ��ҩƷ��ȫ������ҩƷ�İ�ȫ������û��һ�������ľ��Եı���Ҳ����ȷ��һ������ͬ������Ч�ı�Ҳʮ��ģ�����ֽ������ż��ֿ���������1.��LD50����������������С���簴�����ж����LD50��1/6��1/12֮��ļ�����Ϊ��ȫ������
����2.��LD50/ED50��������С��Ч����=����ָ�������Ʊ��ʣ���Ϊ��ȫָ�ꡣ
����3.��LD50������������/ED95��������Ч����=����ָ����
����4.��������С��Ч����/ED95����Ϊ���òο���������������㣩
������ʵ�ϣ���Щҩ�������ָ���dz���խ����ǿ��߰��ҩ�����������ж���֮������С�������ж������������ǽϺõ�ǿ�İ����κ�ҩ�ﶼ�ж����ԣ�ʹ���ʵ��������Ƽ�������֮�����ж�������������˰�ȫ����Եġ���Ӧ������Ч�����������õ�ҩ���ж��٣��Ի��߶��ںͶԺ���Ķ����������ʼ��̶ȵ������ۺϿ��Ƕ��������磬�ֿ���ҩ�ɵ��°�֢�����������ȿ��ΰ����ֿ��°����Ƕ���ѧ�����е����������շ���֢������ȫ�Բ��û�и�ҩ�����������������������г���ͨ����Ӧͣ��������ǧ�����������ͣ�����ȴ�ǿ���財����ѡҩ����������ҽҩ�г���ͨ��
������ҩƷ��Ч�ж�������һ��ҩ���Ƿ���Ч�������ѵ�����Ч�ʿռ���ѡ��ͬ����ϵ����ݲ�һ��ҩ����Ч���۱���һ������磬���ڶ���������Чͨ��ָ���ǣ�����(1)��ȫ���⣺�ɼ���������ȫ��ʧ����һ���¡�
����(2)���ֻ��⣺�������ֱ���������ֱֱ���ij˻���С��50%����������������������һ���¡�
����(3)���⣺���������˻���С����50%��������25%����������һ���¡�
����(4)��չ�����������˻�����25%���ϡ�
�������ڵ�ʵ�尩�ٴ���Ч��Ϊ��
(1)��Ч��������С1/2���ϣ��������Ը��ƣ���Ч����һ�������ϣ����ƺ������䳬�����ꡣ����(2)��Ч��������С����1/2����Ч����һ�������ϡ���������ȶ��������ƣ���Ч�������������ϣ����������䳬������
����(3)��Ч�������״�����ƻ������
��������Ч��Ŀ�У�������Ϊ�����ƺ������䳬�����ꡱ��������Ȼ���㡣��1972�������ͳ��1261���ΰ�����������Ϊ����
�������ʱ��Ϊ3���£�4~6���£�7~9���´�����ֱ�Ϊ1001�ˣ�794�ˣ�624�ˡ������ʷֱ�Ϊ79.4%��59.4%��49.3%����ͳ�ƽ�������ΰ�������7��������ʱ��Ȼ�������Ϊ����.��������ô�����ڰ���ҩ�������ڳ������궩Ϊ��Чָ��ͺܲ����ʡ�
�����ڹ��ڣ���Ϊ������ٴ���Ч��֤ʵ�飬�����ȫ��ͬѧ��Э����������Ʊ��������ںܶಡ���ÿ�ָ����������ð֢״�ĸ��ƣ�ͷ�μ��ᣬ���ʼ��ٵȣ�ʹ��Ч���ж��������ѡ�
����Ҫ֤��ij��ҩ����Ч�ã�Ӧ����ָ��ҩ������еġ����ϵ�ͬһ��Ч��ҩ��Ч���ã���Ч��Ҳ��ָ���ҩ����������������ͬ��λʱ���������ø��á��翹��Ѫѹҩ��ɷֱ�������Ѫ�ܱڡ�������������ĩ�ҡ���-���塢�ں������Ȳ�ͬ��λ��Ҫ�Ƚ����ǵ�������Σ��Ͳ�����ͳ��˵����Ѫѹ��Ρ�
����������ҩ����ٴ�����ָ��ԭ��������ѧԺ���������»ᣨNAS��NRC����Ϊ��ҩƷ����Ӧ��Ϊ�����ȼ���
����(1)��Ч(effective)�������������������µ������е���Ӧ֤���ϡ�
(2)��Ч�����ǣ�effective,but����������ҩ��Ч�����ֳ��ָ��õġ�����ȫ���������ҩ����ԣ�û�������ı�Ҫ������(3)������Ч��probablyeffectivc���������Ͽ���Ч����֤�����Ӳ��㡣
����(4)������Ч��possiblyeffective����֤���ٵ��ֲ���˵�������Ϻ���֤����Ч����ˣ�Ҫ��������Ҫ�IJ������顣
����(5)����ineffectivc��
����(6)�̶�������Ч(ineffectivcinfixcd-combination)����������Ȼ����Чҩ�����û��֤��˵�����õĸ���ҩ�ﶼ���������ã�û��֤�����к��������õij�ּ�����û��֤��˵�������õ�ҩ�ﲻ�����Ӳ��ʵ������á�
������ʵ�ϣ�ҩƷ�İ�ȫ����Ч֮���Ǹ���֤��ϵ��ǿ���κ�һ�涼���С���ȫ����Чֻ����Եģ����Ǿ��Եġ�ҩ�¹�����Ա��������ҩ����������ҩƷ����̭ҩƷʱ����Ҫǡ�������ն���֮��Ĺ�ϵ��
��������ҽԺ�Ƽ������ල
����ҽԺ�Ƽ������ල��ҩƷ���������͡�ҩƷ������ʵʩ�취����ҽ�Ƶ�λ�Ƽ�����Ա���Ƽ��������Լ���ʩ�������������������������˹涨��
1.��Ա����ҽԺ��λ�����䱸����Ӧ��ҩѧ������Ա����ҩѧ������Ա����ֱ�Ӵ���ҩ��������������ҽ�Ƶ�λ�Ƽ���ҩ�츺����Ӧ��ҩʦ���Ϻʹ���ҩ������5�����ϵ�ҩʿ���Ρ�����2.�����빤�ղ��ֹ����Ƽ�����Χ������������Ƽ���Ҫ�����Ƽ���30�����ڲ����й��ޡ���¯����̫ƽ�䡢��Ⱦ�����������ѡ����﷿����ˮ�����Լ�������ȾԴ���Ƽ�����Χ10�����ڲ�����¶�������������ͨ����࣬��ˮ����ͨ���Ƽ����÷��������������Ƽ�Ʒ��Ҫ������Ӧ�����Ƽ�����������֣������������ֿ�����Ϣ�ҡ��칫�����Ƽ��ҷֿ������ơ���װ����ǩ�����װ�ֿ����ڷ��Ƽ��������Ƽ��ֿ�����ͨ�Ƽ�����ҩ�Ƽ���������Ƽ��������Ƽ����ֿ�����ҩ���Ƽ�רû��
�����Ƽ�����ǽ�ڡ�������棨Ӧ����ˮĥʯ���棩����Ӧƽ����࣬����϶������������������ʣ�������ϴ�������������Ƽ�Ҫ������Ӧ��������ȡ�ӡ����¡�������ͨ�缰����������Ⱦ������Ӭ������������������ʩ��Ӧ���������ˮ�Ĵ�ʩ������Ӧ��ˮ����ȡ��Ũ�������������Ȳ�����Ĺ���Ӧ����ͨ�硢��������ʩ��
�����Ƽ�Ӧ���ݼ��͵���Ҫ�����ò����䣬��ÿ�������ֲ�����λ������Һ��⡢��ҺӦ�ڽྻ�������½��С���ע��λ�ྻ����ӦΪ1���ֲ�100�������ϼ侲̬�ⶨӦ�ﵽ�����ˣ����ȸ�λΪ10����Ӧ���ڼ��ྻ������������Ҫ����С���ҩ�Ƽ���Ӧ�ڽྻ���������ơ������Ƽ�Ӧ�ڹ涨�Ľྻ�������Ʊ����ྻ������һ�㷿��Ӧ�л���䣬����������ɵͼ���������������С�
����ƿװ����Ƽ�Ӧ��������ƿ�ӣ����䡢��ѡ�䡢�����䡢��Һ�䡢��ע�䡢����䡢�Ƽ�䡢��װ�䡢�����ȡ���ҩע�������ҩ�Ĵ�������ȡ���䡣
����Ƭ��Ӧ��ԭ����Ԥ�����䡢���顢���ϡ����������������ѹƬ�����¡���װ�����װ���Ƽ����䡣���Ҽ���Ӧ����װ�Ҽ�ͽ�������䡣
���������ڷ�������Һ����ͼ����ࡢ˨����ɢ������������Ӧ���빤��Ҫ������Ӧ�IJ������䡣
�������衢ˮ������Ӧ��ԭ������ѡ��Ԥ���������顢��ɸ�����ۡ����硢���衢�����װ�ȷ��䡣
������ҩ���������������Ӧ��ԭ������ѡ��Ԥ��������ȡ��Ũ������ϡ���������ɡ���װ�ȷ��䣬��������Ӧ�����乤��Ҫ������Ӧ�ķ��䡣
����3.�豸�Ƽ��豸���밴�������̺������֡�����ҩƷֱ�ӽӴ����豸���棬Ӧ��ࡢƽ��������ϴ����ʴ������ҩƷ������ѧ�仯���������ã�����ȾҩƷ���Բ����������ͷ۳����豸��Ӧ�ֱ��ȡ�����������������ų���ʩ��
�������������Ƽ��ͼ�����������DZ�Ӧ��ר�˸������������ά������������Ӧ���Ǽǣ���������������Ҫ����Ӧ�У��豸������������̡�ԭʼ�������ϣ������豸ʹ�á�ά��˵����ԭ������װ��ͼ��ʩ��ͼ����������嵥��ά�ޱ������¹ʼ�¼�ȡ�
�����䱸�����רҵ�����鼰���õIJο����ϣ�����ҩƷ���͵ط�����ҽԺ�Ƽ��淶��ҽԺ�Ƽ����顢ҩ��ѧ���Ƽ�ע�⡢Ӣ������ҽ���ֵ䡢ҩ���ֵ�ȹ����顣
4.�����Ƽ��ұ����������Ҫ������ȫ�����ƶȣ��豸������Ӧ����״̬��ǡ��Ƽ���Ӧ�����������ͨ�����õĸ����Һ������䡣�Ƽ��ұ���ר�õ�һ�㹤�����ͽྻ������������Ьñ�����ֵȣ��������Ե������־����Ӧ����ϴ�����������ڸ���ԱҪ���涨Ҫ��ϴ�֡�ϴ�衢��ָ�ס����¡����ָ���������������û�ױ������������������ô���涨���Ƽ������Ƽ���ԱҪÿ�����һ�Σ���������������������5.�Ƽ����������Ƽ������ƶ����չ�̣����ݰ�����Ʒ��������͡������ɷּ�������������̡������������������ݡ���װҪ��ȡ�����������̽������ƣ���������ı䡣�����Ƽ���ԭ����ҩƷ�������顢���Ӧ����ҩ�������������ϡ���װ��������װ����Ӧ���涨���м����飬���ϱ���ʹ�á��ڷ��Ƽ�����ɫ���ͽ�ζ����Ӧʹ��ʳ��ɫ�غ�ʳ���㾫����ҩ�ġ���ҩ��Ƭ��Ͷ��ǰӦ����Ҫ����α�����Ͻ�Ͷ�ü���ҩƷ��
����������ͨ�Ƽ����õ�����ˮ����ˮ��Ӧ�����й�ҩ��涨��ÿ�����ٽ���һ���ص���Ŀ�ļ��顣
������������Ƽ����õ�ע����ˮ�������Ʊ�����Ӧ�����й�ҩ��涨�����������Ƽ�ǰ��Ӧ��ˮ�ʽ���pHֵ���Ȼ�������ؽ������ص���Ŀ���鲢�м�¼��ÿ�°�ҩ��涨����ȫ����顣
���������Ƽ��밴������Ĺ��չ�̽������ƣ��Բ����Ϲ��չ�̵�ָ������������������ָ��ģ���������Ӧ�ܾ�ִ�С�
��������ÿ��ҩƷ�����ϸ������������������Ĵ�������������Ͷ�ϣ����øı�ɷ֣����õ���Ͷ�ϣ��Ͻ�͵�����ϡ�
�������ƺ�����ҩƷ������ҩƷ������ҩƷ���Ƽ�Ӧ�ϸ�ִ���йع涨��
�����������Ƽ�����ǰ��Ӧ�����峡��������װ�ú��豸��ϴ�ɾ����������Ӧ���ȷ��ǰ���������ϣ����ɿ�ʼ���ơ�
�������ƹ�����ʹ�õ�������������Ŀ�ı�־����������������ơ������Σ��ţ���ת�еĻ����豸��ҲӦ���мӹ��������Ƶı�־�����������ĸ�����ǩ�֡�ÿ���Ƽ���Ӧ��һ���������ط�ӳ����ҩƷ����ȫ���̵�ԭʼ��¼�����ƹ��̺ͽ�������Ƽ����������ǩ�֣��浵3�ꡣ��¼���ݰ���ҩƷ���ơ����͡��������ڡ����š��������Լ���Ʒ���չ����������ƵĹ������̿��������ƹ����н��е�ȫ�������������ۼ�ǩ�֡����ڡ��Ӹ����ŵı�ǩ��
�����Ƽ�������ԱӦ��ʱ��д������¼���ּ��塢������ʵ������ȷ�����ɲ�����Ա��������Աǩ�֡���¼Ӧ�������࣬����˺�ٺ�����Ϳ�ģ�����Ҫ�ģ�Ӧ�ɸ�����ǩ�֣�����ʹ�����ĵIJ��ֿ��Ա��ϡ���ԭ�ϵ���Ʒ��ȫ������ԭʼ��¼������������鵵���������ꡣ
6.�Ƽ����鼰��������ҽ�Ƶ�λ�Ƽ��ҡ�ҩ���ҡ�ֱ��ҩ�����쵼�������Ƽ���ģ������ѧ�����䡢�����䡢����䡢�����۲�䡢����������ʵ���ҵȡ�����ҩ���ұ����䱸���������Ƽ�����Ӧ�ļ����豸�����������ҡ�ʵ���ұ������������ͨ�����ã��������²�Ӧ����ʵ��Ҫ��Ӧ����ˮ�����ۡ��ɹ⡢���µ���ʩ����Ӧ��ר�˹���������Ҫ��Ź��ơ�������̭�����¡�����ʹ�ü�¼��
�����Ƽ��ij�ƷҪ���涨���м��飬�ϸ��ʹ�á�
����ҩ���ұ�������ȫ�ļ��鿨�������¼Ӧ��Ź鵵�����ݰ�������������Դ���������顢�������ݡ����ݴ��������۵�ԭʼ���ϡ�
�������ݼ�����Ӧ���鱨���飬�����ˡ�������ǩ�ֺ���ҩ���Ҹ��������ǩ�֡�ȫ����ԭʼ�����¼�����鱨�浥��������װ���ɲᱣ��3�ꡣ�����¼Ӧ�ּ������������ʵ��������ǩ�֣�����˺�ٺ�����Ϳ�ġ�����Ҫ����ʱ��Ӧ�и�����ǩ��������ʹ�������IJ��ֿ��Ա��ϡ�
����ҩ���Ҷ������Ƶ��Ƽ����뽨�������۲��ƶȣ�ָ��ר�˹������ڹ۲�����ѡȡ�ʵ����������й涨��Ŀ�ļ��飬��Һÿ�¼���һ�Σ������Ƽ�Ʒ��ÿ2�¼��һ�Σ�������д��¼��������3�ꡣ
7.��װ�ͱ�ǩ������װ�ͱ�ǩ���뱨ҽԺҩ���������������������������ű�������ǩӦ��ר�˱��ܣ�ʹ��ʵ�еǼ��ƶȡ�����8.�Ƽ�ԭ���Ϲ����Ƽ�����ԭ�ϡ�����Ӧ����ҩ�ñ���û��ҩ�ñ��ģ�����ȫ���鲢��ҩ�¹���ίԱ������ʹ�á�
��������Ƽ����õ�ԭ��ҩƷ���������ҩ�ñ�����ע���ù���ҩƷ��������������ע��������ϸ��ԭ�ϲ�ҩ�ã���ҵ������ԭ�ϼ���ѧ�Լ������������Ƽ�ԭ�ϡ�
��������Ƽ���ˮ����ʹ�����ʵġ����ϡ��й�ҩ�䡷�涨��ע����ˮ��ÿ��Ҫ���ڽ���һ�������飬������¼��ÿ�������Ƽ�ǰӦ���ص���Ŀ��飨pHֵ����Ρ��Ȼ���ؽ�������
������ͨ�Ƽ��õ�����ˮ��������ϡ��й�ҩ�䡷�涨����ÿ�¶��ڶ�����ˮ����һ��ȫ���飬������¼��ÿ��Ӧ������ˮ���ص���Ŀ��飨pHֵ����Ρ��Ȼ���ؽ�������
9��ʵ�鶯��Ĺ������﷿�����ú��������﷿����ַӦѡ���ܱ��ְ�������ࡢ�������Ӱ��ĵط����������﷿������߳��㡢ͨ�����á��������ࡢ����ˮ�����ǽ��Ҫ������ࡢ��������ǽ���ݶ�������Ŵ���ͨ����Ĺܵ��ȣ�����ž�Ұ�������Ӭ�������溦���롣���ڵ��¶���16-28��C��ʪ��60~80%������60�ֱ����ڡ�
������������Ҫ���ͷ硢�ŷ硢���¼���ů�豸��
��������Ҫ������ϴ��������ʹ�Ұ�Ũ����20PPM���¡�
����������ר�õ����ۡ���ˮ��ʩ����ֹ��ԭ��ɢ��
�����������������������ͬ����ʵ�鶯��ƶ�����������̡��Զ��。������������������ֳ������Ԥ����ʩ��������������������ȷ�Ĺ涨�����������。����ʹ�õ�����
�����ƶ�ʹ�ü�¼��ʹ��ǰӦ�۲��飬ʹ�ú�Ҫ���ƴ��������Զ���ʹ�ü��ظ�ʹ��ʱ��Ӧ����ҩ��Ĺ涨��
���������ϸ�����������ƶȡ�����������ʵ��������Ա����Ʒ�����밴��ͬҪ���������DZ�����ʵ������Ա��δ�������Ͳ�ȡ��ʩ���������ڣ��Լ��ٸ�Ⱦ���ᡣ
10.�ҩƷ�ල����Ҫ�ϸ�ִ�о����IJɹ��ƻ���������֯��Դ����֤ҩƷ��Ӧ�������ٴ���Ҫ��Ҫ�Ծ����ع��Ҽ�ҽԺ���йع涨���������ɣ��Ѻ�ҩƷ�����ء����ɹ����ϸ��ҩƷ����ҩƷ������ҵ����֤����ҩƷ��Ӫ��ҵ����֤����ҩƷ���ĺŵ�ҩƷ�ͷ�������ҩƷ��������������������Ͻ���˽�����й���ҩƷ���ɹ�ҩƷҪ�������ʼ����ߺͼ������ȹ���ԭ��ֹ��ѹ���������ɹ�ҩƷҪ�ο����һ���ҩ���Ʒ�֡�����ҩƷҪ����ԭʼ��Ʊ������д�����������ռ�¼������ͬҩ�������Ա��ͬ���ƶ�Ҫ��������⣬��ͬǩ�֣���ʾ���𣬸������ҩ�����ҵ���е�һ�ж�����ϵ������Э��ҩ�������Ա����ҩ������롢�����������ߡ�ҩƷ������Ӧ�ļල
������һ��ҩƷ������Ӧ�ĸ��������
��������������֯��WHO����ҩ�ﲻ����Ӧ���ֳ�ADR���Ķ����ǣ���Ϊ��Ԥ������ϻ����ƣ�����ʹ��һ����ҩ����������κ��к��ķ�Ԥ�ڵ�ЧӦ����ҩ�ﲻ����Ӧϵָ���������÷�����������£������������ҩ��������к�������ķ�Ӧ��
����ҩƷ������Ӧ��ΪA�ͣ������ͣ���B�ͣ��ʱ��ͣ����ࣺ
����A�Ͳ�����Ӧ����ҩ��������ǿ���¡����ص����Ԥ�⣬һ����ʹ�ü�����������ҩ���йأ�������Ⱥ�з�������ߵ��з��ʵ͡�
����B�Ͳ�����Ӧ������ҩ�������ء�һ����ʹ�ü����أ������ʵͶ������ߡ��������Ӧͨ���ַ�ΪҩƷ�쳣�벡���쳣���֡�ҩƷ�쳣һ���������Ч�ɷַֽ⡢ҩƷ���Ӽ����ܼ���ɫ�ؼ����л��ϳɼ��������������쳣�����Ŵ����ߵ��й�ϵ��
����������ҩ�ﲻ����Ӧ
����1�������������Ƽ�����ҩ����������ijЩ�����Ŀ���ص����á��簢��Ʒͨ�������ڽ��θ�����ζ�������ڸɵȡ���Ϊ�����������������Ƽ�����ͬʱ���ֵģ������丱���ó��������Ա���ġ�
����2�����Է�Ӧ��ȻҲ�dz���ʹ�ü�����������ʹ���ߵ����䡢�����ƶ�������ҩ������������ҩʱ���������ġ����෴Ӧ������Σ���ϴ��ٴ������Ķ��Է�Ӧ�У�
������1��������ϵͳ��Ӧ����ͷʹ��ѣ�Ρ�ʧ�ߡ������������ȣ�
������2����Ѫϵͳ��Ӧ���������ϰ���ƶѪ������Ѫϸ�����ٵȣ�
������3������������״�ʹ���������ܼ��ˡ����㡢Ѫ������ȣ�
������4����Ѫ��ϵͳ��Ӧ����Ѫѹ�½����Ķ����٣�����ʧ���ȡ�
����3.������ӦҲ�Ʊ�̬��Ӧ��ֻ���������ʵIJ��˲��ܳ��֣���ҩ������ء��ٴ������Ĺ�����Ӧ�У�
����(1)ȫ���Է�Ӧ����������ݿˡ�ѪҺ������Ӧ��Ѫ�岡����Ӧ����Ѫ��ϵͳ��Ӧ����������Ӧ����ϵͳ��Ӧ����������Ӧ�Լ����෴Ӧ�ȣ�
����(2)Ƥ����Ӧ����̶���ҩ������Ա�Ƥ��иή����ҩ�����ʱ�ɵ�����������������Ƥ����ҩ������ɺ�����ҩ����Ѫ������ˮ����ҩ�ʪ��Ƥ����ҩ����ͺ�ߺ���֢���ͺ��ҩ���������ҩ��ȡ�
����4.ҩ����������Ҫ�����ڳ���ʹ��������ҩƷ���¡����˵����Ľ���Σ������һ�������ͣҩ���ɽ�ϣ�����ʱӦ��ȡǿ���Խ�ϴ�ʩ��
����5.��ͻ���»����°���Ҫ��ʹ�þ�����ͻ�����������ӵ�ҩ�����¡�Ŀǰ��������������ҩʱ������ҩ����������ͻ�����������ӵ�ҩ�������ʹ�á������ڹ�ȥ����ֶ�����Լ���鲻����ɾ��������������õ�ҩƷ�������г��ϡ���ͨ���ٴ�ʹ�á����ͨ��ҩ��ɸѡ��������ЩҩƷ������̭��
����6.����������Ӧ���Ⱥʧ�������ظ�Ⱦ�����Ե��ȡ�
����������ҩƷ������Ӧ��ⱨ���ƶȽ��������������ѽ�ҩƷ������Ӧ�����ΪҩƷ�ල������������Ҫ���ݡ��������ҹ���ҩƷ������Ӧ�����������ѿ�չ������
����1.��֯����ҩƷ���Ϸ�Ӧ��ⱨ��ϵͳ������
����(1)������ѯ������������ҩƷ����ίԱ���ҩƷ������Ӧ���ίԱ�ᡣ��ίԱ�������������ٴ���ҩ�����ٴ�ҩ�����ٴ�ҩѧ����ҩ��ҩƷ���顢ҩ�������ȸ�����ר����ɣ��乤��ֱ�Ӷ�����������
����(2)ִ�л�����
����1)ҩƷ������Ӧ������ģ��칫�������й�ҩƷ������Ʒ�춨����
����2)ҩƷ������Ӧ���վ���ɸ�ʡ����������ֱϽ�����������֣�������
����2.�������
����(1)ҩƷ������Ӧ���ίԱ��
����1)��������ĵĹ����ƻ���
����2)�������ҩƷ������Ӧ������Ϣ�Ĵ����뷢����
����3)��ʱ�������������ҩƷ��������������棻
����4)�����ҩƷ������Ӧ�����˵Ľ�������ý��顣
����(2)ҩƷ������Ӧ�������
����1)�����������ļƻ��Ͱ��ţ����ҹ���ҩƷ������Ӧ������ҵ������֯������
����2)�ռ������������ࡢ�������������Ը��ص�ҩƷ������Ӧ�����������ϣ�
����3)��ҩƷ��ȫ�Է�����ҩƷ�ල���������ṩ��ѯ��
����4)���ڻ��ڱ�ӡ������ҩƷ������Ӧ�������Ϣ����չ����������ָ���ٴ�������ҩ����ȫ��ҩ��
����5)�����������Ϳ�չ���ʿƼ����鱨�����ȡ�
������3��ҩƷ������Ӧ���վ
����1)�ռ�����ʵ��ת�ݱ�Ͻ���ڵĸ���ԭʼ�����飻
����2)��������ͼ�����ķ�����ҩƷ������Ӧ�����鱨��ͨ����
����3)��ɼ�����Ľ���ĸ�������
����3.ҩƷ������Ӧ�������ҽ��������λ��ҽʦ��ҩʦ����ʦ�����ν���������ҩƷ������Ӧ���������ñ����飬���͵���ʡ����������ֱϽ�����������֣��ļ��վ�����б�Ҫ�����ֱ���ϱ�������ġ�
������ҩƷ��������Ӫ��ҵӦע���ռ�ҩƷ������Ӧ��������ʱ����վ���档
(��)����ƶ�����������ʡ����������ֱϽ��������ҽ�Ƶ�λ����ҽҩԺУ�ĸ���ҽԺ��ָ��������ҽԺΪ�ҹ����ص��챨�浥λ�������е�ҩƷ������Ӧ�ı��浥λ�����뼰ʱ��������д��ҩƷ������Ӧ�����顱�ñ������Ϊ���ƻ�ɫ��Ƭ��
�����Է���ҩƷ������Ӧ�Ĵ���
(1)��һ���������в�ѧ���������ʵ���о�������(2)�����ٴ�ҽ��ע�⣬����ʹ�û��鳧����˵���飻
����(3)������������ͣ����ʹ�á�����ص��챨�浥λ����ϵͳ���졣
����(4)��ֹ������ֹͣ����ʹ�ã�ֱ����̭��
����(��)���ͼ��������������������ṩ����Ϣȷ��������ҩ��ȫ������Ч���������乱�״�С������������������Ա����߸��轱���ͱ��ã�����Ϊרҵ����ְ������IJο���
��������ҩƷ����Ԥ��IJ�����Ӧ�������غ���ģ�ֱ���й���Աδ�豨���������������ߣ�Ӧ��Υ����ҩƷ������������ʮ�����ڶ���涨���д���������������Ӧ����ǹ������Ĺ涨����Ԥ��ģ�����ù�صĹ������飩�����ڸ��˹�ʧ������������غ�������˻��²С���������Ӧ���ա�ҩƷ������������ʮ�����涨�������º���λ���߸���Ӧ�������⳥���Ρ��ܺ��˿��������ؼ����������������Ŵ������ܺ���Ҳ����ֱ��������Ժ���ߡ�
������ҩ�ﲻ����Ӧ�ļ�ⱨ�淶Χ����1.����Σ���������²�ֱ��ɥʧ�Ͷ������������IJ�����Ӧ��
����2.��ҩͶ��ʹ�ú���ĸ��ֲ�����Ӧ��
����3.��ΪҩƷ���µ�ͻ�䡢���䡢���Σ�
����4.�������͵Ĺ�����Ӧ��
����5.������ҩƷ������ҩ�������ԣ�
����6.��ΪҩƷ������õ��µIJ�����Ӧ��
����7.����һ����ҩƷ�йص�����IJ�����Ӧ��
�����ˡ�ҩѧ�ල�������뷽��
�������ҹ���ҩƷ�ල������ͨ��������Ŀ�������ʵ�ֵġ�Ŀ�����ͨ��5Wԭ�����£�what������ʱ��when����˭ȥ�죨who���ε�(where)��������(how)��ͨ��ȷ��Ŀ�ꡢ�ƶ��ƻ���ʵʩ�ල���Ƶȹ����ֶΣ��ﵽ������Ŀ�ģ����������GMP��GSP��GLP��Ӧ��Ŀ������ľ������á�ҽԺҩѧ�ලĿ����������ݣ�Ҫ���ݡ�ҩƷ����������ҩ��ҩ�棬ȷ��������Ŀ�꣬������ȫ��������ƶȣ��ƶ��ල���˱���������֯ʵʩҽԺҩѧ����������ҩ���������۱�����ʵʩ�����в��ϼල��飬���۹���������ʵ��ҽԺҩѧ�������淶�����ƶȻ���ѧ�������μ��ھ��£���
�������£�