�����ڡ�Ⱦɫ������ۺ���
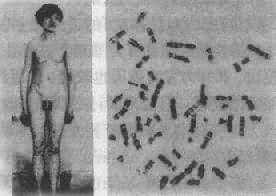
����Ⱦɫ�岡��chromosomal disease����Ⱦɫ������ۺ�����chromosome aberration syndrome ����һ�������ص��Ŵ�����ͨ�����з������κ��������£�ͬʱҲ�ǵ��������벻������Ҫԭ��һ�����Ⱦɫ��������0.5%-0.7%�Ļ��Ӥ����7.5%��̥�����Է�������Լ1��2��Ⱦɫ���쳣���ֽ���֪��Ⱦɫ�岡����100�֣��ѱ����Ⱦɫ����Ŀ�ͽṹ�쳣��500�����ϡ����Ÿ߷ֱ��Դ�������ϸ���Ŵ�ѧ�¼�����Ӧ�ã���ᷢ�ָ����Ⱦɫ�岡���쳣��
����һ��Ⱦɫ������ۺ����ĸ��
����Ⱦɫ������ۺ�����ָ����Ⱦɫ���쳣������ļ������������ж����ٴ����֣��ʳ�Ϊ�ۺ�����ͨ�����û��Ⱦɫ�����������������١���һЩȾɫ������(ƽ����λ����λ)�Ͳ�һ����������쳣��Ⱦɫ��Ķ�̬����̬�ԣ�polymorphism��heteromorphism��ͨ���������쳣���ͣ��ʲ���ΪȾɫ������ۺ�����
��������Ⱦɫ���쳣������Ƶ��
�����ۺ�������ҵ����ϣ���Լ��15���������������������һ��ΪȾɫ���쳣���£���ԼΪ5��-8������̥��Ⱦɫ���쳣�������ڳ���ǰ��90������������Ȼ�������������������磬��Ⱦɫ���쳣��Ƶ�����ߡ�������Ⱦɫ���쳣����������2��3��
������ͬ����Ⱦɫ���쳣������Ƶ����ز�������0.47-0.84%֮�䣬�ñ�2��3�еķ����ʶ��ҹ���������Ⱦɫ���쳣�������������ƹ��㣨��2��4����
������ͨ����Ⱦɫ���������Ϻ��١�1986-1987�꣬�ҹ��Ĵ�ʡ�����й����ģ���Ŵ������в�ѧ�������飬��Ⱦɫ�岡���ߵĻ��������2��5��
��2��3 56 952��������ϸ���Ŵ�ѧ�����
Ⱦɫ���쳣���� |
�쳣���� |
�� |
���Ʒ����� |
��Ⱦɫ�壭���� |
98 |
0��260 |
1/400 |
47��XXY |
35 |
0��093 |
1/1 000 |
47��XYY |
35 |
0��093 |
1/1 000 |
���� |
28 |
0��074 |
1/1 300 |
��Ⱦɫ�壭Ů�� |
29 |
0��151 |
1/700 |
45��X |
2 |
0��010 |
1/10 000 |
47��XXX |
20 |
0��014 |
1/1 000 |
���� |
7 |
0��037 |
1/3 000 |
��Ⱦɫ�������� |
82 |
0��144 |
1/700 |
��D |
3 |
0��005 |
/20 000 |
+ E |
7 |
0��120 |
1/8 000 |
+ G |
71 |
0��125 |
1/800 |
���� |
1 |
0��002 |
1/50 000 |
ƽ��Ľṹ���� |
110 |
0��193 |
1/500 |
��ƽ��ṹ���� |
34 |
0��597 |
1/2 000 |
�ܼ� |
353 |
0��620 |
1/160 |
��2��4 �ҹ�������Ⱦɫ���쳣�����Ĺ���
�쳣���� |
�쳣Ƶ�ʣ� |
����ֵ�������꣩ |
��Ⱦɫ���쳣 |
0.223 |
46138 |
��Ⱦɫ����Ŀ�쳣 |
0.144 |
29793 |
Ⱦɫ��ṹ���� |
�� | �� |
ƽ��� |
0.193 |
39793 |
��ƽ��� |
0.060 |
12414 |
�ܼ� |
0.620 |
128276 |
(��1981������˿����Ƽ���)
��2��5 �Ĵ�ʡ��ͨ��Ⱥ��Ⱦɫ�岡�Ļ�����
| ���� | ������ |
| 21�������� | 0��14 |
| ������Ⱦɫ�岡 | 0��02 |
| �������ѳ�������ȫ | 0��07 |
| ������غ�跢����ȫ | 0��07 |
| ����Ⱦɫ���쳣 | 0��015 |
| �ܼ� | 0��315 |
*������غ�跢����ȫ��������Ϊɸ�����Ѷ���ֵƫ��
����������Ⱦɫ���쳣�ۺ�֤
������һ�������ۺ���
����1���������� ��������������Ҫ��Ⱦɫ�弲����Ӣ��ҽ��Langdon Down �����������ʳ�Ϊ Down�ۺ�����Down sydrome����1959�꣬����ϸ���Ŵ�ѧ��Lejeune֤ʵ�˲��IJ����Ƕ���һ��С��G��Ⱦɫ�壨����ȷ��Ϊ21�ţ����ʴ˲��ֳ�Ϊ21�����ۺ����� Lejeune�ķ��ֿ�����ҽѧ�Ŵ�ѧ��һ����Ҫ��֧�D�D�ٴ�ϸ���Ŵ�ѧ��
������1�������ʣ���������21�����ۺ����ķ�����ԼΪ1��800��1.25���������Ի�������Ů�ԡ�ĸ��������Ӱ�췢���ʵ���Ҫ���ء����ݹ������ϣ����һ���˳���ʱ��ĸ������ƽ��28.2�꣬������ĸ��ƽ������Ϊ34.4�꣬��ĸ��20���Ϊ1��2000��35���Ϊ1��300��40���Ϊ1��100��45�������1��50��
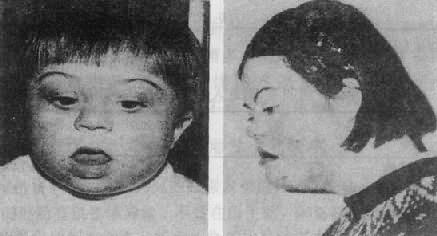
������2���ٴ����֣��������ͻ�������ʱ���غ�����ƫ�ͣ����������£�ͻ�������沿���Σ�ͼ2��17����ͷС��Բ��������ƽ����Բ����ƽ���DZ�ƽ,����ϸ��������б���۾����������Ƥ���ԣ�����б�ӣ���Ĥʱ�аװߵ㣬������״����ǣ���С����������죨�������մ�֮���ɴ˶���������С����λ�ͣ��������Σ��������̶������ж����Ƥ�����������Ƿ����������֫�϶̣��ֿ����ʣ�ͨ���ƣ�ָ�̣���5ָ�����䣬��С��ȱСָ�нڣ�Ƥ��Ҳ��һ�����ص㣨���ĵ�ʮ�£��������������¶����ͣ��ʳ��и�ֱ����������ޣ�Լ1��2���ϵĻ��������������ಡ����Ҫ���Ҽ��ȱ�𡢷��ҵ���ͨ������������δ��,�������Ļ�����ʮ��ָ������խ���᳦��ֱ���Ѵ������ű�����Ҳż���ɼ��������Գ�����غ��غ�����������̣������ӳ����٣������½�����δ���������ߡ�Ů�Ի���ͨ�����¾���������������������������������ͻ��������£�mental retardation,MR���DZ�����ͻ�������صı��֣�����̶��ڸ����߲���ȫ��ͬ������ͨ����25-50֮�䣬����50�ĺ��١���Ϊ�����������ڶ��ͻ�������˼ά�����������
������3��ʵ���Ҽ�飺��������᪻�ø��SOD��1�����Կ�����50������ø����λ21q22�������л������ЧӦ�����⣬���߶���Ʒ�ر����У�������������ȱ�ݻ������Խ����������¡�Ӧ��������ص�֢״�������߹���ʧ�������ܰ�ϸ���ͱ��������ǻ�����Ⱦ��ԭ��
ͼ2��17���������ͻ���
��ͼ��1��Ů��
��ͼ��16��Ů��
������4�����ͣ����Ϳɷ�Ϊ���ͣ����͵ı����ǣ����͵ģ������ͣ�����47����21ռ95����Ƕ���ͼ�46��47����21ռ1��-2%;��λ��ռ3��-4����������ȫ����ϸ������һ��21��Ⱦɫ�壬�ٴ�֢״���Ͷ�������������Ƕ����ͨ����������ϸ��ϵ����֢״����ȡ�����쳣ϸ����ռ�ı������ʲ���ܴ�һ��ϵ�����Ϊ�ᡣ�������ϸ�����٣�����������������졣��λ�͵ĺ����ж���,�������Dq21q��ռȫ����λ�͵� 54.2%,�����21qGq,ռ40.9����������λ��5����һ��˵������λ�͵�֢״�ȵ��͵�Ҫ��Щ����Dq21q�У��������14q21q��ռDq21q��58.5%�����Ϊ13q21q��ռ22%����15q21qռ19.5%��21qGq��λ�У�21q21qռ83.3%����21q22q��ռ16.6%�����������λ��������Ȼֻ��46���壬����һ��21����λ������һ��D���G��Ⱦɫ���ϣ���������������21�ţ��Զ���һ�������21�ų��ۣ������������Ĺؼ�����Ϊ21�ų��ۣ����ٴ����Ա��ֳ�21�����֢״��
������5���Ŵ�ѧ�����͵�21���弸�������·�����de novo���ģ��븸ĸ�ĺ����أ����Ǽ�������ʱ������Ľ�����������볣������ĸ����ֳϸ����Լռ��������95������5�����ڸ�����������Ҫ�����ڵ�һ�μ������ѡ����͵�21����ֻ�м���һ�������Ŵ��ģ���ĸ���DZ������ߡ����⣬�����ų�ijЩ����������ĸ��ʵ����21����ϸ�����ٵ�Ƕ���壬������ǵ���Ů�п��ܻ�ö����21��Ⱦɫ�塣���Ի��߲���������û���Ŵ�����һ�������⡣
����Ƕ���ͻ������������������ϵ�ϸ��ϵ��ͼ2��6���������Ǻ��Ӻ�post-zygotic����˿���Ѳ�����Ľ���������һ������ʱ���������룬�ͻ����47����21��45����21����ϸ��ϵ������һ��ϸ���Ǻ�ά���ġ���ˣ�����Ƕ����IJ������뷢�����Ժ��ij����˿���ѣ�����Ƕ�����ڶ���������ϸ��ϵ��
������λ�͵�21����������ϸ������һ����λ��Ⱦɫ�壬����ͨ����һ����������Ⱦɫ����21��Ⱦɫ�峤��ͨ����˿���ںϣ�������λ�����ɡ�Dq21q��λ�У�55%���·����ģ�45%������˫��֮һ��ƽ����λ��21qGq ��λ����ȫ��(96%)���·����ģ����Ŵ������Ľ�ռ4����
����������λ���Ŵ������ͬ��Dq21qƽ����λ��Я����ͨ���������ѿ����γ�6�����ӣ����ܾ�������ܷ������⣬���Բ�������̥������λ�����廼����ƽ����λЯ��������̥����ͼ2��12������ˣ����ƽ����λЯ���ߵ�˫������Ҫ���塣
����21qGq��λ�У�21q22q��21q21q��λ���Ŵ�ѧ���岻��ȫ��ͬ�����˫��֮һΪ21q21ƽ����λЯ���ߣ���û�п����������������̥������Ϊ����ֻ�ܲ����������ĺ��ӡ�21q22q��λ���Ŵ������Dq22q���ƣ�ֻ��ǰ�߸���ͨ�����״��ݶ����߶���ĸ�״��ݵ�����
������6���Ŵ���ѯ�����������ٷ����ղ�ͬ���������͵�21�����ԣ��ٷ�����������������������������35������ԼΪ0.5%��35��������ԼΪ1������Ȼ������˫�ף�ͨ����ĸ�ף�֮һǶ��������ջ����ӡ���֮���Ѳ���һ��21���廼�������ٷ�����0.5%,��Ⱦɫ�������ܷ���Ϊ1.2���������ų�����������Ⱦɫ���쳣�Ļ�����
������λ���д�Լ1��2�IJ������·����ģ���1��2��˫��֮һƽ����λ����ġ�ǰ�߸������պ�С����ƽ����λ���µ��ٷ���������Ը��ݾ�����ơ���������˫��֮һΪƽ����λЯ���ߣ�������������Ϊ33.3%����ʵ�ʷ��յ͵ö࣬����˫����һ��ΪЯ�����йء�Dq21q��λЯ��������ĸ�ף����������ķ���Ϊ10��-15������Ϊ���ף������Ϊ5�����С��21q21q��λ�������֮������ͬ������λȾɫ���ɸ������ݵİٷֱȽ�D��G��λҪ�࣬��������10�����¡�21q21qƽ����λЯ���ߵĺ��100������������������Я���߲���������
������7��Ԥ����ƽ������ֻ��16.2�ꡣ50���Ļ�����5����ǰ������ֻ��8���Ļ��߳���40�꣬2.6%����50�ꡣ�����Ĵ�ʡ�����ϣ���Ⱥ�л����ʽ�ԼΪ����ʱ��1��10��
����2��13�����ۺ��� 1960��Patau�����������������ֳ�ΪPatau�ۺ������������еķ�����ԼΪ1��25 000��Ů�����Զ������ԡ�
������1���ٴ����֣������Ļ��κ��ٴ�����Ҫ��21���������صöࣨͼ2��18������Ļ��ΰ���Сͷ��ǰ�ǰ�Է���ȱ�ݣ�����С�����к�Ĥȱ�𣬱ǿ�����ƽ��2��3�������ϴ��ѣ����������ѣ���λ�ͣ��������Σ��С������������ָ��ֺ������ָ��ǵ���������ͻ�������������γ���νҡ�ε��㡣���Գ������һ��κ���غ��Ů���������ٷʴ�˫������˫���ӹ��ȡ��Ժ�����Ļ��ηdz��ձ飬�������ԣ����һ��ķ����ȱ�𡢶�������δ�գ������������ۻ�ˮ�ȣ������ڶ�������ȱ����ɶ�����
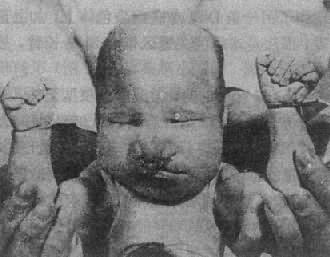
ͼ2��18 Patau�ۺ�������ʾ����ֵĻ���
�������������ϰ��������еĻ��ߣ����ҳ̶����أ����ϾõĻ���������������������Ź������µȡ�
������2��ϸ���Ŵ�ѧ���Ŵ���ѯ��80���IJ���Ϊ������13���壬����Ϊ46��XX����XY������13���������ΪǶ���ͻ���λ�͡�Ƕ����һ��֢״���ᣬ��λ��ͨ����13��14��������λ�Ӷ࣬������һ��t(13q14q)��λȾɫ�壬����Ϊ46����14����t(13q14q)�������Ƕ���һ��13�ų��ۣ���˫��֮һ��ƽ����λЯ����ʱ����Ϊ��������쳣̥�����������������������ķ��ղ�����5����1�������˫��֮һΪ13q13q��λЯ���ߣ�����ֻ�ܲ����������ĺ��ӣ������ʴ�100��
������3������Ԥ����13�� �巢�ͱ��ص�������֪���٣�ĸ���������ԭ��֮һ������ĸ��ƽ������Ϊ31.6�꣬����ƽ������Ϊ34.6�ꡣ���⣬�����ϱ�����79�����������ں��伾�ڣ�9��2�£���45���Ļ����ڳ�����һ������������90����6�����������������3��������5����ƽ������Ϊ130�졣
����3��18�����ۺ�����1960��Edward���������������ֳ�ΪEdward�ۺ���(Edward syndrome ).18�����Ե������ػ��Σ��ڳ�����������������Լ1��3500��8000������������ijЩ�������������ߣ��ﵽ1��450��800��������Ů�ԣ����Ա�Ϊ4��1��
������1���ٴ����֣���������ʱ���صͣ�ƽ����2243g,���������������˱���Ӧ����ͷ�沿�����������ػ��Σ�ͼ2��19����ͷ��������������Բ���۾����������Ƥ������С����Ĥ���ǣ�����ϸ������С����λ�ͣ��������Σ�������������Сmicrognathia��,���̣��ж����Ƥ����ȫ�������������쳣���عǶ̣�������խ�����ɹ��ޣ���ֱ������ȡ��ֵĻ��ηdz����ͣ�����ȭ��Ĵָ���������ָ�ϣ�������ָ������ǣ�ָ������ȫ����ָ�����ƹ��࣬Լ1��3����Ϊͨ���ơ���֫��ͻ�����ǡ�ҡ�ε��㡱��Ĵֺ�̣�����������ֳ�����αȽϳ���������غ������������ٷ��������ȡ�95���IJ��������������ಡ�����Ҽ��ȱ�𡢶�������δ�յȣ�������������Ҫԭ�������Σ����ۻ�ˮҲ�ܳ�������������������ȱ�ݣ�������ʱ��̣ܶ��������Բ�����

ͼ2��19 18�����ۺ������ߣ����沿���������ң��ֵĵ�����ȭʽ
������2��ϸ���Ŵ�ѧ��80�����ߺ���Ϊ47��XY����XX������18����10������ΪǶ���壬��Ϊ46��XY����XX����47��XY����XX������18������Ϊ������λ����Ҫ��18����D��Ⱦɫ�����λ��˫����ƽ����λЯ���߶�����18�������ߺ��١�
������3��Ԥ���������2��3������������ƽ�����71�죬ֻ�м������˳�����ͯ�ڡ�Ƕ���͵Ĵ���ڱȽϳ���
����4������Ⱦɫ�������ۺ��� �Ƚ���Ҫ����8�š�22�������ۺ����ȡ����������Եķ������κ��������¡�����һϵ������λ����Ⱦɫ�岿�������ۺ��������ٴ�֢״ȡ���ڶ���Ⱦɫ��Ƭ�ε����ʺʹ�С��Ⱦɫ�岿�������Կɷ�Ϊ�����ࣺһ����ijһȾɫ��Ƭ�ε������ԣ��ظ�����ͬʱ�ְ�������Ⱦɫ����쳣����ȱʧ����λ������һ�ಿ�������Եı��ͱȽϸ��ӣ��������ظ���ȱʧƬ�ε�ijЩ֢״����һ��ΪȾɫ���ijһƬ�εĵ����ظ��������ԣ��������༫Ϊ�ټ���
����(��)���弰���ֵ����ۺ���
����������Ⱦɫ��Ķ�ʧͨ���������ģ������Ϊ��������ȷ��СȾɫ�壨��21�ţ���ȫ��ʧ�ı��档������λ�����γɻ�ȱʧ���µ�Ⱦɫ�岿�ֵ�����Ƚ϶��������5q���ۺ�������è���ۺ���Ϊ�������¡�
����è���ۺ�����5q���ۺ�����Ϊ�����ȱʧ�ۺ������䷢���ʹ���Ϊ1��50000��Ů�Զ������ԡ���Ӥ�Ŀ����dz���Сè�����������ʵ����������沿���ƺܻ��飬��ʵ���������·dz����أ����̳�����20������������Ҳ�����ԡ��������ٴ����ֻ���Сͷ�������������ѹ���������Ƥ�����С�Һ�����ͼ2��20����Լ20�����������������ಡ����Ҫ���Ҽ��ȱ��Ͷ�������δ�յȡ�
�������ߵ�Ⱦɫ��ȱʧƬ�δ�С��һ��֢״��Ҫ��5p15��ȱʧ���𡣻���������·����ġ���Ⱦɫ��Ƭ�εĵ���ȱʧ�������м�ȱʧ����ռ80������ƽ����λ�����ռ10������״Ⱦɫ���Ƕȫ����Ƚ��ټ������״�Ⱦɫ�����ŵ��µ�5p���ۺ����������
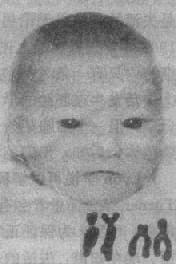
ͼ2��20è���ۺ��������䲿�ֺ���
�������ߵ������ʵͣ������ܻ���ꡣ
��2��6 �״�ƽ����λЯ�����ٳ�����ƽ����λ�����ĸ��ʣ�����
����� |
��֤�� |
�״�Я���� |
|
ĸ |
�� |
||
2ƽ������λ |
��ƽ�� |
15 7 |
|
ƽ���������λ |
ƽ�� |
�� |
|
D��G������λ |
��ƽ�� |
15 7 |
|
D��G������λ |
ƽ�� |
�� |
|
D��G������λ |
ƽ���ƽ�� |
��С |
|
������Jacobs P A.1979��
�����ġ���Ⱦɫ���쳣�ۺ���
������һ���Ա����Ⱦɫ��
����1���Ա�ľ��� ������X��Y������Ⱦɫ�壬����������ı����Ա���YȾɫ�塣���ٴ��Ͽ������������⣬����Y�߽�Ϊ���ԣ������ΪŮ�ԡ���ΪYȾɫ�����о���غ�跢����غ��������ӣ�testis determining factor,TDF������غ��Ĵ��ھ����˸�����Ա�
����2��XȾɫ���ʧ�� Ů��������XȾɫ�������ֻ��һ������Ů��XȾɫ��Ļ�����ﲢδ�����Զ�1����������ŮX�������������ȵ��������Ŵ�ѧһ��Ϊ����������dosage compensation��������һ����Ľ����ǣ���ȻŮ��������XȾɫ�壬������һ����ʧ��ģ����������Ů��ֻ��һ���й��ܵ�XȾɫ�塣Ӣ���Ŵ�ѧ��Mary Lyon ��1961���������������XȾɫ��ʧ���˵����Lyon��˵����Ҫ���ǣ��ٴ��Բ��鶯��ϸ����ֻ��һ��XȾɫ���л��ԣ���һ��ʧ������������ڼ���ϸ������Ϊ��Ⱦɫ�ʣ���ʧ�������̥�����ڣ���ʧ��������ģ���ʧ���XȾɫ��ȿ����Ը���Ҳ������ĸ�ף���һ��ϸ��ij��X����ʧ��ɸ�ϸ�����ܶ�������ϸ��������ͬһ��ʧ���XȾɫ�塣��֪��XȾɫ��ʧ����������ڣ�Լ������16�����ҡ�
����Lyonѧ˵���Խ��������Ŵ����������Lyon��˵���ܽ��ͺ��Ժ���ΪXO��Turner�ۺ��������и����쳣���ֺ��Զ�X�������и���֢״������XԽ��֢״Խ���ء��ɼ���Ϊ��֤�����ķ�������������̥������ijһʱ����Ҫ˫��XȾɫ���ϵĻ�������֪����ʧ���һ��XXȾɫ���ϵĻ���ȫ��ʧ��ⷽ������һЩ֤�ݡ�����֪XgѪ�ͻ���Ѱ��ţƤѢ������Dz�ʧ��ġ������������YȾɫ����һЩ��XȾɫ���ͻ���ͬԴ�Ļ����������������Ի�Ů�Զ��������������XO����ȱ��һ�ݶ�XXX���������ݣ���֮���б����쳣��
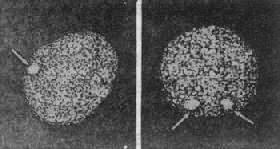
����ʧ���XȾɫ���ڼ��ڳʹ���״̬����ΪXȾɫ�ʣ�X��chromatin�����ڿ�ǻ��ճĤϸ��������ϸ���ж������ü�Ⱦɫ���������ͼ2��21����XȾɫ�ʵ���Ŀ��XȾɫ����Ŀ��1����������������XȾɫ���쳣ʱ������ͨ������Ⱦɫ�ʼ������������ϡ����磬XȾɫ����XOʱΪ0������Ů��Ϊ1��XXY����Ϊ1��XXX����Ϊ2��
������������Ⱦɫ����Ŀ�쳣�ۺ���
����1��Klinefelter�ۺ�����Klinefelter syndrome���ֳ�Ϊ������غ�跢����ȫ��ԭ��Сغ��֢��������Ⱦɫ��ΪXXY�������������Զ���һ��XȾɫ�壬��֮���ೣ��ΪXXY�ۺ�����
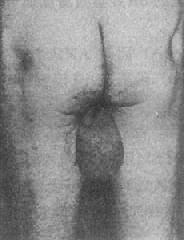
����Klinefelter�ۺ����ķ������൱�ߣ������������дﵽ1.2�롣���ݰ����˵����ϣ�����180cm�����Ի�����Ϊ1��260���ھ������������ݻ�����Ϊ1��100������������������ԼΪ1��20���ٴ�����Ϊغ��С����Ӳ����ϸ����ή�����ʲ������䡣�������Ӳ�������97�����߲������������Եڶ������������Ů�Ի����֣������룬��ë�٣���ë�ֲ���Ů�ԣ�������ͷС�ȣ�Լ25���Ļ������鷿������ͼ2��22�����������ĸߡ���֫����һ���ֻ��ߣ�Լ1��4�����������£�һЩ�����о����쳣�����������֢����ʵ���Ҽ��ɼ��Ƽ������࣬19������ͪ���ߣ����ص�ʧ���뻼�ߵ�Ů�Ի������йء�
����
����
ͼ2��21��XȾɫ�ʡ�ͼ2��22 Klinefelter�ۺ������������
��������������ߵĺ���Ϊ47��XXY����Լ��15������Ϊ���������ϸ��ϵ��Ƕ���壬���г�����Ϊ46��XY��47��XXY;46,XY��48��XXXY�������X�������״���������ʱXȾɫ�岻����Ľ����
������غ��ͪ���ƿ����յ�һ����Ч�������ɴ�ʹ�ڶ������������Բ����ߵ�����״̬��
����2��XYY�ۺ��� ����Ӥ�еķ�����Ϊ1��900��XYY���Եı��͵������ģ��������ĸߴ�����180cm��ż���ɼ���غ��غ�跢����ȫ���о������ϰ����������½���������ѵȣ�����������Կ���������XYY���������˷ܣ��е����������㣬��ѧ�����ҿ�������ײ�����������Ϊ��XYY�����Ǹ������γɹ����еڶ��μ�������ʱ����YȾɫ�岻����Ľ����
����3��Turner�ۺ��� �ֳ�Ϊ45��X��45��XO�ۺ�����Ů�����������ٷ�����ȫ���������ѳ�������ȫ�ۺ�����������ŮӤ�еķ�����ԼΪ0.2��-0.4�룬�����Է�������̥��Turner�ۺ����ķ����ʿɸߴ�7.5��.���߱���ΪŮ�ԣ����İ�С������һ������������������ͬ������������Σ��������´�������Ƥ�ȣ����ͻխ�����С�Һ������ڽ��������������죬�����ķ��ʺܵͣ���һֱ���ӵ��粿��Լ50���������뾱�����������״Ƥ����˫�羶�����ؿ�ƽ��ܣ���ͷ�����ٷ��������ͷ��������ⷭ�ڱ���ʮ�ֵ��ͣ�ͼ2��23�������ġ������ƹǶ̶���

ͼ2��23 Turner�ۺ������������
�����䣬������ָ������ȫ��Ӥ���ڽű����ܰ����ף�ʮ�����⡣������ֳϵͳ���쳣��Ҫ���ѳ��������״���٣����������γɣ��ӹ�������ȫ������ԭ���Ավ�����������ѳ����ܵ��»��ߵ���ëϡ�٣���Ҹë������ֳ�����ɡ����⣬��Լ��1��2��������������խ���������Ȼ��Ρ�
����Turner�ۺ����ĺ��ͳ����͵�45��X��Լռ55�����⣬���и���Ƕ���ͺͽṹ�쳣�ĺ��͡��������Ƕ����46��XX��45��X��46��X,i��Xq����һ��˵����Ƕ���͵��ٴ����ֽ��ᣬ����YȾɫ���Ƕ���Ϳɱ��ֳ����Ի������������İ�С������Turner֢״��Ҫ����X�̱۵����Ծ����ģ����ѳ�������ȫ�벻��������볤�۵������йء�
����Turner�ۺ����ķ���������˫�������γɹ����еIJ����룬����Լ75����Ⱦɫ�嶪ʧ�����ڸ�����Լ10���Ķ�ʧ�����ں��Ӻ���������ʱ��
���������������������ػ�����������������֮�⣬һ����ܴ�ֻ�����ഺ�ڲű�����������������ϰ�Ҳ���ᣬӦ�ü�����14����ǰ��ʼ���ƿ��Դٽ��ڶ���������ֳ���ٵķ������¾�����������״̬�ı䣬�����ܴٽ����ߣ������߿�������
����4��47��XXX��XŮ�� �����ֳ�Ϊ���ƣ�superfemale����������ԼΪ0.8���1/2250��
����������������XȾɫ���Ů���������Ρ��Թ��������������������ģ�ֻ�������������¾����١��̷��վ���������������Լ��2��3�������Եͣ����л���������
��������47��XXX�⣬һЩ���ߵĺ���ΪǶ���ͣ���47��XXX��46��XX��XXX���ߵ�ĸ����������ƽ��Լ����4�ꡣ�������������Ҫ������ĸ��һ��������������4������5��XȾɫ�塣һ����˵��XȾɫ�����࣬�����ͷ������������ء�
������������Ⱦɫ��Ľṹ����
����1��XȾɫ��Ľṹ�쳣 ������XȾɫ��ṹ�쳣�и���ȱʧ����λ�͵ȱ�Ⱦɫ�塣���ǵ��ٴ����ֶ�������Ҫȡ�����漰XȾɫ���ϵ���Щ�����쳣����Ϊ��ͬ���������еĻ���ͬ��ȱʧ���µ�֢״Ҳ��ͬ��Wyss������һTurner�ۺ����ĺ�����֢״��ϵͼ���������˽�XȾɫ��ṹ�쳣�ı��֣�ͼ2��24����

ͼ2��24 Turner�ۺ���������֢״���ͼ
������1��X�̱�ȱʧ��XXp-����XpԶ��ȱʧ�������������İ�С��Turner�ۺ��������������ٹ���������Xpȱʧ����������̱ۣ�������Turner�ۺ������������������ٷ�����ȫ��XȾɫ�峤�۵�Ⱦɫ��[X,i,(Xq)]���ٴ�����������ƣ���ΪҲȱʧ�������̱ۡ�
������2��X����ȱʧ��XXq-����ȱʧ��q22��Զ�ߣ�һ��������ٷ�����ȫ��ԭ���վ������������������������İ�С��Turner�ۺ���������ȱʧ��Χ�ϴ������۽����ߣ������ٷ�����ȫ�⣬һЩ��������۴���������XȾɫ��ȱ�Ⱦɫ��[Xi(p)]������ơ�Xq�м�ȱʧ�ۼ�q13-q26�����ٹ������������������������ɼ��ж�ȱʧ��Turner���������йء�
����ͨ������ȱʧ`�γɻ�״��ȱ�Ⱦɫ���X��ѡ���Ե�ʧʡ�£��Ӷ���֤��һ��������X��
������3����λ����XȾɫ���볣Ⱦɫ�巢��ƽ����λʱ�����ڻ���ƽ��ı��֣�һ�㲻�����֢״����ʱʧ���������XȾɫ�塣����ƽ����λ�ϵ���q12-q26ʱ���л��Ե�X�ڸ�������Ϊ�����֣��ͻᵼ�����ٷ����쳣�����⣬�糣Ⱦɫ��ڶ���λ��XȾɫ�������ƽ����λʱ����������˫��˿��Ⱦɫ�壬�����ȡ����Xp��Xq�϶��ѵ��λ�á�
����2������XȾɫ���ۺ��� �����ͳ�һЩѧ��ע������»��������Զ���Ů�ԡ�1943��Martin��Bell��һ����ϵ�������з���11�����Ի��ߺ���������������µ�Ů�ԣ���Ϊ�ü�ϵ������������X�����ģ����X�������������ֳ�ΪMartin-Bell�ۺ�����
����1969��Lubs�����������������»�����Ů�������з����˳��۾��С�����ͳ�ϸ˿״���˺ۡ���XȾɫ�塣������Sortherland ֤��ϸ˿λλ��XȾɫ�峤��2��7����Xq27�������ڵ�Ҷ�����������±��������˴��Բ�λ��franile site���ĸ���ֽ����ǰ���Xq27���д��Բ�λ��XȾɫ���ΪXȾɫ�壨fragile X,fra X����ͼ2-25�������������µļ�����Ϊ����XȾɫ���ۺ�����
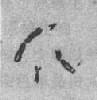

ͼ2-25 ����XȾɫ�壨Fra X���ۺ���
��ͼ������XȾɫ��
��ͼ�����ߴ��`���
��ͼ�����ߴ�غ��
������1�������ʣ������������еķ�����Ϊ1/1000~1/1500���������������͡������������������»���Լ10%~20~Ϊ����������
������2���ٴ����֣���Ҫ����Ϊ�жȵ��ضȵ��������£����������������������������س����������������죬ǰ��ͻ�������в�������ȫ�������ǰͻ����������������´�ͻ������һ����Ҫ�ı����Ǵ�غ��֢��һЩ�����жද֢����������Ϊ����֢��20%��������������ȥ����Ϊ����Ů��������XȾɫ�壬���Ů��Я���߲��ᷢ��������������XȾɫ������һ��ʧ�Ů���Ӻ�����Լ1/3��������������¡�
������3�������ķ��ӻ������ֽ���X���Բ����ѷ������²�����FMR-1�������У�CGC��n ���˸����ظ����У�������������ԼΪ30�����������������Դ����ߺ�Ů��Я�������ൽ150~500bp����ΪС���룬���ڵ�Cpg ��δ������������ǰͻ�䣨premutation ����ֻ����֢״��Ů��Я���ߵ�CGG�����ȶ����������ۺ�����ݹ��������������������Ի��ߺʹ��Բ�λ�߱����Ů�Դﵽ1000~3000bp�����ڵ�CpG��Ҳ������������ȫͻ�䣨full mutation���ɹر����ڻ���ı���Ӷ������ٴ�֢״����ǰͻ��ת��Ϊ��ȫͻ��ֻ����ĸ���������ݹ����С����ݶԴ��Բ�λDNA���е��˽⣬���ѿ���RFLP����������DNA�ӽ�������PCR�����ȷ���������²�����
������4�����ƣ�Lejeune ��ΪҶ��ȱ����Fra X�ۺ���ʱ�������µ�ԭ�����ô����Ҷ�����ƻ���������õ�Ч��������������δ��֤ʵҶ�����Ч���½�һЩ������Ϊ�������˷ܼ���Ч�Ϻã��������ô��������ÿ��ֶ���clonidine�����ĵð��ߣ��ݳƿɼ���ද֢��
����3��YȾɫ�弰��ṹ�쳣 YȾɫ����ӫ��Ⱦɫʱ����δ���п����ļ�������ӫ���������21��22������YȾɫ�峤�۵Ķ�̬�Էdz����ԣ�����������죬��Y���й��˺��ձ����еı����ϸߡ�
����4��YȾɫ�����Ŀ�쳣 ����ǰ��������XYY�Լ���Y�ۺ�����XXYY��X-
-�ȣ���YȾɫ��Ľṹ�쳣����Y�ij��ۻ�̱�ȱʧ���ȱ�Ⱦɫ��i(Yq)��i��Yp������״Ⱦɫ���˫��˿��Ⱦɫ�壨Ϊ����Y�Ķ̱ۡ� ����������Y�ij������ںϣ�����λ�����漰Y����λ����Y�볣Ⱦɫ�壬Y��XȾɫ�����λ�ȣ��������Է�ת�ۺ���46��XX������Yq�ϵ�SRY������λ��һ��XȾɫ�����£���46��XYŮ����SRY����ȱʧ��ͻ��Ľ����
����˼�٣�